Analysis of colorectal adenocarcinomas
Giulio Caravagna
Source:vignettes/Cross_CRC_MSeq.Rmd
Cross_CRC_MSeq.RmdData
Cross et al. The evolutionary landscape of colorectal tumorigenesis. Nat Ecol Evol. 2018 2(10). PMID 30177804.
-
n=19adenomas (unmatched); - multi-region sequencing with binary data;
- annotated driver events from Cross et al.;
Building the cohort
data(CROSS_CRC_ADENOCARCINOMA_NATECOEVO_2018, packages = 'evoverse.datasets')
print(CROSS_CRC_ADENOCARCINOMA_NATECOEVO_2018)
#> # A tibble: 63 × 7
#> patientID variantID CCF is.clonal is.driver Misc cluster
#> <chr> <chr> <chr> <lgl> <lgl> <chr> <chr>
#> 1 adenoma_1 ACVR2A R1:1;R2:1;R3:1;R4:1;R5… TRUE TRUE NOTH… 1
#> 2 adenoma_1 APC R1:1;R2:1;R3:1;R4:1;R5… TRUE TRUE NOTH… 1
#> 3 adenoma_2 APC R1:1;R2:1;R3:1;R4:1 TRUE TRUE NOTH… 1
#> 4 adenoma_2 KRAS R1:0;R2:0;R3:0;R4:1 FALSE TRUE NOTH… 2
#> 5 adenoma_2 ARID2 R1:0;R2:0;R3:0;R4:1 FALSE TRUE NOTH… 2
#> 6 adenoma_2 TP53 R1:1;R2:1;R3:1;R4:0 FALSE TRUE NOTH… 3
#> 7 adenoma_3 PIK3CA R1:0;R2:0;R3:0;R4:1;R5… FALSE TRUE NOTH… 1
#> 8 adenoma_3 FBXW7 R1:1;R2:1;R3:1;R4:1;R5… TRUE TRUE NOTH… 2
#> 9 adenoma_3 APC R1:1;R2:1;R3:1;R4:1;R5… TRUE TRUE NOTH… 2
#> 10 adenoma_3 AKAP9 R1:0;R2:0;R3:1;R4:0;R5… FALSE TRUE NOTH… 3
#> # ℹ 53 more rows
# Constructor
CROSS_CRC_ADENOCARCINOMA_REVOLVER = revolver_cohort(
CROSS_CRC_ADENOCARCINOMA_NATECOEVO_2018,
MIN.CLUSTER.SIZE = 0,
annotation = "Colorectal adenocarcinomas (Cross et al, PMID 30177804)")
#> [ REVOLVER ~ Cohort constructor ]
#>
#> ℹ Using only driver mutations.
#> ℹ Rejecting clusters with less than 0 mutations.
#>
#> ── REVOLVER input data ─────────────────────────────────────────────────────────
#>
#> # A tibble: 63 × 9
#> Misc patientID variantID cluster is.driver is.clonal CCF id
#> <chr> <chr> <chr> <chr> <lgl> <lgl> <chr> <chr>
#> 1 NOTHING adenoma_1 ACVR2A 1 TRUE TRUE R1:1;R2:1;R3:1… __mu…
#> 2 NOTHING adenoma_1 APC 1 TRUE TRUE R1:1;R2:1;R3:1… __mu…
#> 3 NOTHING adenoma_2 APC 1 TRUE TRUE R1:1;R2:1;R3:1… __mu…
#> 4 NOTHING adenoma_2 KRAS 2 TRUE FALSE R1:0;R2:0;R3:0… __mu…
#> 5 NOTHING adenoma_2 ARID2 2 TRUE FALSE R1:0;R2:0;R3:0… __mu…
#> 6 NOTHING adenoma_2 TP53 3 TRUE FALSE R1:1;R2:1;R3:1… __mu…
#> 7 NOTHING adenoma_3 PIK3CA 1 TRUE FALSE R1:0;R2:0;R3:0… __mu…
#> 8 NOTHING adenoma_3 FBXW7 2 TRUE TRUE R1:1;R2:1;R3:1… __mu…
#> 9 NOTHING adenoma_3 APC 2 TRUE TRUE R1:1;R2:1;R3:1… __mu…
#> 10 NOTHING adenoma_3 AKAP9 3 TRUE FALSE R1:0;R2:0;R3:1… __mu…
#> # ℹ 53 more rows
#> # ℹ 1 more variable: cluster_size <int>
#>
#> ── Preprocessing data (this may take some time)
#>
#> ...................
#>
#> ── Extracting clones table ─────────────────────────────────────────────────────
#> → adenoma_1 : 2 entries, 1 clone(s).
#> → adenoma_2 : 4 entries, 3 clone(s).
#> → adenoma_3 : 8 entries, 4 clone(s).
#> → adenoma_4 : 5 entries, 3 clone(s).
#> → adenoma_5 : 3 entries, 1 clone(s).
#> → adenoma_6 : 2 entries, 1 clone(s).
#> → adenoma_7 : 3 entries, 1 clone(s).
#> → adenoma_8 : 4 entries, 1 clone(s).
#> → adenoma_9 : 1 entries, 1 clone(s).
#> → carcinoma_1 : 3 entries, 2 clone(s).
#> → carcinoma_10 : 1 entries, 1 clone(s).
#> → carcinoma_2 : 5 entries, 1 clone(s).
#> → carcinoma_3 : 1 entries, 1 clone(s).
#> → carcinoma_5 : 2 entries, 1 clone(s).
#> → carcinoma_6 : 5 entries, 1 clone(s).
#> → carcinoma_7 : 4 entries, 1 clone(s).
#> → carcinoma_8 : 2 entries, 1 clone(s).
#> → carcinoma_9_distal : 3 entries, 1 clone(s).
#> → carcinoma_9_proximal : 5 entries, 1 clone(s).We can check the cohort, and flag put drivers that are not recurrent.
# Diagnostic
revolver_check_cohort(CROSS_CRC_ADENOCARCINOMA_REVOLVER)
#> ┌──────────────────────────────────────────────────────────────────────┐
#> │ │
#> │ WARNING - Driver variantIDs occuring only once could be removed. │
#> │ │
#> └──────────────────────────────────────────────────────────────────────┘
#> # A tibble: 9 × 7
#> variantID numClonal p_clonal numSubclonal p_subclonal N_tot p_tot
#> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
#> 1 ACVR2A 1 0.0526 0 0 1 0.0526
#> 2 TGIF1 1 0.0526 0 0 1 0.0526
#> 3 SMAD3 1 0.0526 0 0 1 0.0526
#> 4 SOX9 1 0.0526 0 0 1 0.0526
#> 5 ARID2 0 0 1 0.0526 1 0.0526
#> 6 AKAP9 0 0 1 0.0526 1 0.0526
#> 7 GNAS 0 0 1 0.0526 1 0.0526
#> 8 SMAD4 0 0 1 0.0526 1 0.0526
#> 9 CHD4 0 0 1 0.0526 1 0.0526
#> ┌───────────────────────────────────────────────────────────────────────────────────────────┐
#> │ │
#> │ WARNING - Some patients have only one clone with drivers; they will just be expanded. │
#> │ │
#> └───────────────────────────────────────────────────────────────────────────────────────────┘
#> # A tibble: 15 × 7
#> patientID numBiopsies numMutations numDriverMutations numClonesWithDriver
#> <chr> <int> <int> <int> <int>
#> 1 adenoma_1 6 2 2 1
#> 2 adenoma_5 4 3 3 1
#> 3 adenoma_6 2 2 2 1
#> 4 adenoma_7 2 3 3 1
#> 5 adenoma_8 2 4 4 1
#> 6 adenoma_9 2 1 1 1
#> 7 carcinoma_10 5 1 1 1
#> 8 carcinoma_2 7 5 5 1
#> 9 carcinoma_3 6 1 1 1
#> 10 carcinoma_5 6 2 2 1
#> 11 carcinoma_6 13 5 5 1
#> 12 carcinoma_7 8 4 4 1
#> 13 carcinoma_8 5 2 2 1
#> 14 carcinoma_9_… 5 3 3 1
#> 15 carcinoma_9_… 5 5 5 1
#> # ℹ 2 more variables: numTruncalMutations <int>, numSubclonalMutations <int>
# Driver events that occur in 1 patient
non_recurrent = Stats_drivers(CROSS_CRC_ADENOCARCINOMA_REVOLVER) %>%
filter(N_tot == 1) %>%
pull(variantID)
# Remove drivers
CROSS_CRC_ADENOCARCINOMA_REVOLVER = remove_drivers(CROSS_CRC_ADENOCARCINOMA_REVOLVER, non_recurrent)
#> ── Removing driver events ──────────────────────────────────────────────────────
#>
#> # A tibble: 9 × 7
#> variantID numClonal p_clonal numSubclonal p_subclonal N_tot p_tot
#> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
#> 1 ACVR2A 1 0.0526 0 0 1 0.0526
#> 2 TGIF1 1 0.0526 0 0 1 0.0526
#> 3 SMAD3 1 0.0526 0 0 1 0.0526
#> 4 SOX9 1 0.0526 0 0 1 0.0526
#> 5 ARID2 0 0 1 0.0526 1 0.0526
#> 6 AKAP9 0 0 1 0.0526 1 0.0526
#> 7 GNAS 0 0 1 0.0526 1 0.0526
#> 8 SMAD4 0 0 1 0.0526 1 0.0526
#> 9 CHD4 0 0 1 0.0526 1 0.0526
#> ℹ Retained 19 patients after driver removal..
#> ┌───────────────────────────────────────────────────────────────────────────────────────────┐
#> │ │
#> │ WARNING - Some patients have only one clone with drivers; they will just be expanded. │
#> │ │
#> └───────────────────────────────────────────────────────────────────────────────────────────┘
#> # A tibble: 16 × 7
#> patientID numBiopsies numMutations numDriverMutations numClonesWithDriver
#> <chr> <int> <int> <int> <int>
#> 1 adenoma_1 6 2 1 1
#> 2 adenoma_5 4 3 3 1
#> 3 adenoma_6 2 2 2 1
#> 4 adenoma_7 2 3 3 1
#> 5 adenoma_8 2 4 4 1
#> 6 adenoma_9 2 1 1 1
#> 7 carcinoma_1 4 3 2 1
#> 8 carcinoma_10 5 1 1 1
#> 9 carcinoma_2 7 5 4 1
#> 10 carcinoma_3 6 1 1 1
#> 11 carcinoma_5 6 2 2 1
#> 12 carcinoma_6 13 5 5 1
#> 13 carcinoma_7 8 4 3 1
#> 14 carcinoma_8 5 2 2 1
#> 15 carcinoma_9_… 5 3 3 1
#> 16 carcinoma_9_… 5 5 4 1
#> # ℹ 2 more variables: numTruncalMutations <int>, numSubclonalMutations <int>Constructing mutation trees
revolver uses the mtree package to implement
mutation trees from binary data.
# Compute the trees
CROSS_CRC_ADENOCARCINOMA_REVOLVER = compute_mutation_trees(CROSS_CRC_ADENOCARCINOMA_REVOLVER)
#>
#> =-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-
#> Constructing Mutations Tree objects via mtree - https://caravagn.github.io/mtree/
#> =-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-
#> Input patients.
#> adenoma_1, adenoma_2, adenoma_3, adenoma_4, adenoma_5, adenoma_6, adenoma_7, adenoma_8, adenoma_9, carcinoma_1, carcinoma_10, carcinoma_2, carcinoma_3, carcinoma_5, carcinoma_6, carcinoma_7, carcinoma_8, carcinoma_9_distal, carcinoma_9_proximal
#> Warning: replacing previous import 'igraph::%s%' by 'matrixcalc::%s%' when
#> loading 'mtree'
#> Warning: replacing previous import 'igraph::groups' by 'tidygraph::groups' when
#> loading 'mtree'
#> [ mtree ~ generate mutation trees for adenoma_1 ]
#> Sampler : 10000 (cutoff), 5000 (sampling), 100 (max store)
#> Suppes' conditions : >=
#> # A tibble: 1 × 10
#> cluster nMuts is.driver is.clonal R1 R2 R3 R4 R5 R6
#> <chr> <int> <lgl> <lgl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
#> 1 1 2 TRUE TRUE 1 1 1 1 1 1
#>
#> Sampler: this model has 1 node, it has trivial models.
#> ✔ Trees with non-zero sscore 1 storing 1
#>
#>
#> Combinations of Information Transfer : 0
#> [ mtree ~ generate mutation trees for adenoma_2 ]
#> Sampler : 10000 (cutoff), 5000 (sampling), 100 (max store)
#> Suppes' conditions : >=
#> # A tibble: 3 × 8
#> cluster nMuts is.driver is.clonal R1 R2 R3 R4
#> <chr> <int> <lgl> <lgl> <dbl> <dbl> <dbl> <dbl>
#> 1 2 2 TRUE FALSE 0 0 0 1
#> 2 1 1 TRUE TRUE 1 1 1 1
#> 3 3 1 TRUE FALSE 1 1 1 0
#>
#> There are no alternatives!
#>
#> Warning: `progress_estimated()` was deprecated in dplyr 1.0.0.
#> ℹ The deprecated feature was likely used in the mtree package.
#> Please report the issue to the authors.
#> This warning is displayed once per session.
#> Call `lifecycle::last_lifecycle_warnings()` to see where this warning was
#> generated.
#> ✔ Trees with non-zero sscore 1 storing 1
#>
#> Combinations of Information Transfer : 0
#> [ mtree ~ generate mutation trees for adenoma_3 ]
#> Sampler : 10000 (cutoff), 5000 (sampling), 100 (max store)
#> Suppes' conditions : >=
#> # A tibble: 4 × 9
#> cluster nMuts is.driver is.clonal R1 R2 R3 R4 R5
#> <chr> <int> <lgl> <lgl> <dbl> <dbl> <dbl> <dbl> <dbl>
#> 1 2 5 TRUE TRUE 1 1 1 1 1
#> 2 1 1 TRUE FALSE 0 0 0 1 1
#> 3 3 1 FALSE FALSE 0 0 1 0 0
#> 4 4 1 FALSE FALSE 0 1 0 0 0
#>
#> There are no alternatives!
#> ✔ Trees with non-zero sscore 1 storing 1
#>
#> Combinations of Information Transfer : 0
#> [ mtree ~ generate mutation trees for adenoma_4 ]
#> Sampler : 10000 (cutoff), 5000 (sampling), 100 (max store)
#> Suppes' conditions : >=
#> # A tibble: 3 × 8
#> cluster nMuts is.driver is.clonal R1 R2 R3 R4
#> <chr> <int> <lgl> <lgl> <dbl> <dbl> <dbl> <dbl>
#> 1 1 3 TRUE TRUE 1 1 1 1
#> 2 2 1 TRUE FALSE 0 0 1 0
#> 3 3 1 FALSE FALSE 0 0 0 1
#>
#> There are no alternatives!
#> ✔ Trees with non-zero sscore 1 storing 1
#>
#> Combinations of Information Transfer : 0
#> [ mtree ~ generate mutation trees for adenoma_5 ]
#> Sampler : 10000 (cutoff), 5000 (sampling), 100 (max store)
#> Suppes' conditions : >=
#> # A tibble: 1 × 8
#> cluster nMuts is.driver is.clonal R1 R2 R3 R4
#> <chr> <int> <lgl> <lgl> <dbl> <dbl> <dbl> <dbl>
#> 1 1 3 TRUE TRUE 1 1 1 1
#>
#> Sampler: this model has 1 node, it has trivial models.
#> ✔ Trees with non-zero sscore 1 storing 1
#>
#>
#> Combinations of Information Transfer : 0
#> [ mtree ~ generate mutation trees for adenoma_6 ]
#> Sampler : 10000 (cutoff), 5000 (sampling), 100 (max store)
#> Suppes' conditions : >=
#> # A tibble: 1 × 6
#> cluster nMuts is.driver is.clonal R1 R2
#> <chr> <int> <lgl> <lgl> <dbl> <dbl>
#> 1 1 2 TRUE TRUE 1 1
#>
#> Sampler: this model has 1 node, it has trivial models.
#> ✔ Trees with non-zero sscore 1 storing 1
#>
#>
#> Combinations of Information Transfer : 0
#> [ mtree ~ generate mutation trees for adenoma_7 ]
#> Sampler : 10000 (cutoff), 5000 (sampling), 100 (max store)
#> Suppes' conditions : >=
#> # A tibble: 1 × 6
#> cluster nMuts is.driver is.clonal R1 R2
#> <chr> <int> <lgl> <lgl> <dbl> <dbl>
#> 1 1 3 TRUE TRUE 1 1
#>
#> Sampler: this model has 1 node, it has trivial models.
#> ✔ Trees with non-zero sscore 1 storing 1
#>
#>
#> Combinations of Information Transfer : 0
#> [ mtree ~ generate mutation trees for adenoma_8 ]
#> Sampler : 10000 (cutoff), 5000 (sampling), 100 (max store)
#> Suppes' conditions : >=
#> # A tibble: 1 × 6
#> cluster nMuts is.driver is.clonal R1 R2
#> <chr> <int> <lgl> <lgl> <dbl> <dbl>
#> 1 1 4 TRUE TRUE 1 1
#>
#> Sampler: this model has 1 node, it has trivial models.
#> ✔ Trees with non-zero sscore 1 storing 1
#>
#>
#> Combinations of Information Transfer : 0
#> [ mtree ~ generate mutation trees for adenoma_9 ]
#> Sampler : 10000 (cutoff), 5000 (sampling), 100 (max store)
#> Suppes' conditions : >=
#> # A tibble: 1 × 6
#> cluster nMuts is.driver is.clonal R1 R2
#> <chr> <int> <lgl> <lgl> <dbl> <dbl>
#> 1 1 1 TRUE TRUE 1 1
#>
#> Sampler: this model has 1 node, it has trivial models.
#> ✔ Trees with non-zero sscore 1 storing 1
#>
#>
#> Combinations of Information Transfer : 0
#> [ mtree ~ generate mutation trees for carcinoma_1 ]
#> Sampler : 10000 (cutoff), 5000 (sampling), 100 (max store)
#> Suppes' conditions : >=
#> # A tibble: 2 × 8
#> cluster nMuts is.driver is.clonal R1 R2 R3 R4
#> <chr> <int> <lgl> <lgl> <dbl> <dbl> <dbl> <dbl>
#> 1 1 2 TRUE TRUE 1 1 1 1
#> 2 2 1 FALSE FALSE 1 0 0 0
#>
#> There are no alternatives!
#> ✔ Trees with non-zero sscore 1 storing 1
#>
#> Combinations of Information Transfer : 0
#> [ mtree ~ generate mutation trees for carcinoma_10 ]
#> Sampler : 10000 (cutoff), 5000 (sampling), 100 (max store)
#> Suppes' conditions : >=
#> # A tibble: 1 × 9
#> cluster nMuts is.driver is.clonal R1 R2 R3 R4 R5
#> <chr> <int> <lgl> <lgl> <dbl> <dbl> <dbl> <dbl> <dbl>
#> 1 1 1 TRUE TRUE 1 1 1 1 1
#>
#> Sampler: this model has 1 node, it has trivial models.
#> ✔ Trees with non-zero sscore 1 storing 1
#>
#>
#> Combinations of Information Transfer : 0
#> [ mtree ~ generate mutation trees for carcinoma_2 ]
#> Sampler : 10000 (cutoff), 5000 (sampling), 100 (max store)
#> Suppes' conditions : >=
#> # A tibble: 1 × 11
#> cluster nMuts is.driver is.clonal R1 R2 R3 R4 R5 R6 R7
#> <chr> <int> <lgl> <lgl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
#> 1 1 5 TRUE TRUE 1 1 1 1 1 1 1
#>
#> Sampler: this model has 1 node, it has trivial models.
#> ✔ Trees with non-zero sscore 1 storing 1
#>
#>
#> Combinations of Information Transfer : 0
#> [ mtree ~ generate mutation trees for carcinoma_3 ]
#> Sampler : 10000 (cutoff), 5000 (sampling), 100 (max store)
#> Suppes' conditions : >=
#> # A tibble: 1 × 10
#> cluster nMuts is.driver is.clonal R1 R2 R3 R4 R5 R6
#> <chr> <int> <lgl> <lgl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
#> 1 1 1 TRUE TRUE 1 1 1 1 1 1
#>
#> Sampler: this model has 1 node, it has trivial models.
#> ✔ Trees with non-zero sscore 1 storing 1
#>
#>
#> Combinations of Information Transfer : 0
#> [ mtree ~ generate mutation trees for carcinoma_5 ]
#> Sampler : 10000 (cutoff), 5000 (sampling), 100 (max store)
#> Suppes' conditions : >=
#> # A tibble: 1 × 10
#> cluster nMuts is.driver is.clonal R1 R2 R3 R4 R5 R6
#> <chr> <int> <lgl> <lgl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
#> 1 1 2 TRUE TRUE 1 1 1 1 1 1
#>
#> Sampler: this model has 1 node, it has trivial models.
#> ✔ Trees with non-zero sscore 1 storing 1
#>
#>
#> Combinations of Information Transfer : 0
#> [ mtree ~ generate mutation trees for carcinoma_6 ]
#> Sampler : 10000 (cutoff), 5000 (sampling), 100 (max store)
#> Suppes' conditions : >=
#> # A tibble: 1 × 17
#> cluster nMuts is.driver is.clonal R1 R10 R11 R12 R13 R2 R3
#> <chr> <int> <lgl> <lgl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
#> 1 1 5 TRUE TRUE 1 1 1 1 1 1 1
#> # ℹ 6 more variables: R4 <dbl>, R5 <dbl>, R6 <dbl>, R7 <dbl>, R8 <dbl>,
#> # R9 <dbl>
#>
#> Sampler: this model has 1 node, it has trivial models.
#> ✔ Trees with non-zero sscore 1 storing 1
#>
#>
#> Combinations of Information Transfer : 0
#> [ mtree ~ generate mutation trees for carcinoma_7 ]
#> Sampler : 10000 (cutoff), 5000 (sampling), 100 (max store)
#> Suppes' conditions : >=
#> # A tibble: 1 × 12
#> cluster nMuts is.driver is.clonal R1 R2 R3 R4 R5 R6 R7
#> <chr> <int> <lgl> <lgl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
#> 1 1 4 TRUE TRUE 1 1 1 1 1 1 1
#> # ℹ 1 more variable: R8 <dbl>
#>
#> Sampler: this model has 1 node, it has trivial models.
#> ✔ Trees with non-zero sscore 1 storing 1
#>
#>
#> Combinations of Information Transfer : 0
#> [ mtree ~ generate mutation trees for carcinoma_8 ]
#> Sampler : 10000 (cutoff), 5000 (sampling), 100 (max store)
#> Suppes' conditions : >=
#> # A tibble: 1 × 9
#> cluster nMuts is.driver is.clonal R1 R2 R3 R4 R5
#> <chr> <int> <lgl> <lgl> <dbl> <dbl> <dbl> <dbl> <dbl>
#> 1 1 2 TRUE TRUE 1 1 1 1 1
#>
#> Sampler: this model has 1 node, it has trivial models.
#> ✔ Trees with non-zero sscore 1 storing 1
#>
#>
#> Combinations of Information Transfer : 0
#> [ mtree ~ generate mutation trees for carcinoma_9_distal ]
#> Sampler : 10000 (cutoff), 5000 (sampling), 100 (max store)
#> Suppes' conditions : >=
#> # A tibble: 1 × 9
#> cluster nMuts is.driver is.clonal R1 R2 R3 R4 R5
#> <chr> <int> <lgl> <lgl> <dbl> <dbl> <dbl> <dbl> <dbl>
#> 1 1 3 TRUE TRUE 1 1 1 1 1
#>
#> Sampler: this model has 1 node, it has trivial models.
#> ✔ Trees with non-zero sscore 1 storing 1
#>
#>
#> Combinations of Information Transfer : 0
#> [ mtree ~ generate mutation trees for carcinoma_9_proximal ]
#> Sampler : 10000 (cutoff), 5000 (sampling), 100 (max store)
#> Suppes' conditions : >=
#> # A tibble: 1 × 9
#> cluster nMuts is.driver is.clonal R1 R2 R3 R4 R5
#> <chr> <int> <lgl> <lgl> <dbl> <dbl> <dbl> <dbl> <dbl>
#> 1 1 5 TRUE TRUE 1 1 1 1 1
#>
#> Sampler: this model has 1 node, it has trivial models.
#> ✔ Trees with non-zero sscore 1 storing 1
#>
#>
#> Combinations of Information Transfer : 0
#> Fitting models with REVOLVER
Function revolver_fit implements the 2-steps REVOLVER
algorithm to fit the data.
We use the following parameters:
-
initial.solution = NA, to sample random initial solutions for every run of EM; -
n = 3, to repeat the fit 3 times, and retain the one with lower median goodness-of-fit penalty. -
parallel = FALSE, to run serially the fits;
CROSS_CRC_ADENOCARCINOMA_REVOLVER = revolver_fit(
CROSS_CRC_ADENOCARCINOMA_REVOLVER,
parallel = F,
n = 3,
initial.solution = NA)
#> [ REVOLVER Transfer Learning fit ~ Colorectal adenocarcinomas (Cross et al, PMID 30177804) ]
#> ┌───────────────────────────────────────────────────────────────────────────────────────────┐
#> │ │
#> │ WARNING - Some patients have only one clone with drivers; they will just be expanded. │
#> │ │
#> └───────────────────────────────────────────────────────────────────────────────────────────┘
#> # A tibble: 16 × 7
#> patientID numBiopsies numMutations numDriverMutations numClonesWithDriver
#> <chr> <int> <int> <int> <int>
#> 1 adenoma_1 6 2 1 1
#> 2 adenoma_5 4 3 3 1
#> 3 adenoma_6 2 2 2 1
#> 4 adenoma_7 2 3 3 1
#> 5 adenoma_8 2 4 4 1
#> 6 adenoma_9 2 1 1 1
#> 7 carcinoma_1 4 3 2 1
#> 8 carcinoma_10 5 1 1 1
#> 9 carcinoma_2 7 5 4 1
#> 10 carcinoma_3 6 1 1 1
#> 11 carcinoma_5 6 2 2 1
#> 12 carcinoma_6 13 5 5 1
#> 13 carcinoma_7 8 4 3 1
#> 14 carcinoma_8 5 2 2 1
#> 15 carcinoma_9_… 5 3 3 1
#> 16 carcinoma_9_… 5 5 4 1
#> # ℹ 2 more variables: numTruncalMutations <int>, numSubclonalMutations <int>
#>
#> Fitting N = 19 patients
#>
#> # A tibble: 19 × 6
#> patientID hasTrees numTrees maxScore minScore combInfTransf
#> <chr> <lgl> <int> <dbl> <dbl> <int>
#> 1 adenoma_1 TRUE 1 1 1 1
#> 2 adenoma_2 TRUE 1 0.0113 0.0113 1
#> 3 adenoma_3 TRUE 1 0.0000852 0.0000852 1
#> 4 adenoma_4 TRUE 1 0.00255 0.00255 1
#> 5 adenoma_5 TRUE 1 1 1 1
#> 6 adenoma_6 TRUE 1 1 1 1
#> 7 adenoma_7 TRUE 1 1 1 1
#> 8 adenoma_8 TRUE 1 1 1 1
#> 9 adenoma_9 TRUE 1 1 1 1
#> 10 carcinoma_1 TRUE 1 0.0505 0.0505 1
#> 11 carcinoma_10 TRUE 1 1 1 1
#> 12 carcinoma_2 TRUE 1 1 1 1
#> 13 carcinoma_3 TRUE 1 1 1 1
#> 14 carcinoma_5 TRUE 1 1 1 1
#> 15 carcinoma_6 TRUE 1 1 1 1
#> 16 carcinoma_7 TRUE 1 1 1 1
#> 17 carcinoma_8 TRUE 1 1 1 1
#> 18 carcinoma_9_distal TRUE 1 1 1 1
#> 19 carcinoma_9_proximal TRUE 1 1 1 1
#>
#> Initial solution : Randomized (uniform probability)
#>
#> Sampled solutions: n = 3
#>
#> Parallel exectuion (via 'easypar') : TRUE
#> [1] "w"
#>
#> =-=-=-=-=-=-=-=-=-=-=-=-=-=-=-
#> 1] Expectation Maximization
#> =-=-=-=-=-=-=-=-=-=-=-=-=-=-=-
#>
#> Number of Solutions
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Combinations of Transfer 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
#>
#> Initialization 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
#>
#> # 1 : E: OK M: 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |# A tibble: 19 × 9
#> patientID hasTrees numTrees maxScore minScore combInfTransf Solution
#> <chr> <lgl> <int> <dbl> <dbl> <int> <int>
#> 1 adenoma_1 TRUE 1 1 e+0 1 e+0 1 1
#> 2 adenoma_2 TRUE 1 1.13e-2 1.13e-2 1 1
#> 3 adenoma_3 TRUE 1 8.52e-5 8.52e-5 1 1
#> 4 adenoma_4 TRUE 1 2.55e-3 2.55e-3 1 1
#> 5 adenoma_5 TRUE 1 1 e+0 1 e+0 1 1
#> 6 adenoma_6 TRUE 1 1 e+0 1 e+0 1 1
#> 7 adenoma_7 TRUE 1 1 e+0 1 e+0 1 1
#> 8 adenoma_8 TRUE 1 1 e+0 1 e+0 1 1
#> 9 adenoma_9 TRUE 1 1 e+0 1 e+0 1 1
#> 10 carcinoma_1 TRUE 1 5.05e-2 5.05e-2 1 1
#> 11 carcinoma_10 TRUE 1 1 e+0 1 e+0 1 1
#> 12 carcinoma_2 TRUE 1 1 e+0 1 e+0 1 1
#> 13 carcinoma_3 TRUE 1 1 e+0 1 e+0 1 1
#> 14 carcinoma_5 TRUE 1 1 e+0 1 e+0 1 1
#> 15 carcinoma_6 TRUE 1 1 e+0 1 e+0 1 1
#> 16 carcinoma_7 TRUE 1 1 e+0 1 e+0 1 1
#> 17 carcinoma_8 TRUE 1 1 e+0 1 e+0 1 1
#> 18 carcinoma_9_distal TRUE 1 1 e+0 1 e+0 1 1
#> 19 carcinoma_9_proxi… TRUE 1 1 e+0 1 e+0 1 1
#> # ℹ 2 more variables: converged <lgl>, penalty <dbl>
#>
#> =-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-
#> 2] Transfering orderings across patients
#> =-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-
#> [1] "w"
#>
#> =-=-=-=-=-=-=-=-=-=-=-=-=-=-=-
#> 1] Expectation Maximization
#> =-=-=-=-=-=-=-=-=-=-=-=-=-=-=-
#>
#> Number of Solutions
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Combinations of Transfer 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
#>
#> Initialization 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
#>
#> # 1 : E: OK M: 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |# A tibble: 19 × 9
#> patientID hasTrees numTrees maxScore minScore combInfTransf Solution
#> <chr> <lgl> <int> <dbl> <dbl> <int> <int>
#> 1 adenoma_1 TRUE 1 1 e+0 1 e+0 1 1
#> 2 adenoma_2 TRUE 1 1.13e-2 1.13e-2 1 1
#> 3 adenoma_3 TRUE 1 8.52e-5 8.52e-5 1 1
#> 4 adenoma_4 TRUE 1 2.55e-3 2.55e-3 1 1
#> 5 adenoma_5 TRUE 1 1 e+0 1 e+0 1 1
#> 6 adenoma_6 TRUE 1 1 e+0 1 e+0 1 1
#> 7 adenoma_7 TRUE 1 1 e+0 1 e+0 1 1
#> 8 adenoma_8 TRUE 1 1 e+0 1 e+0 1 1
#> 9 adenoma_9 TRUE 1 1 e+0 1 e+0 1 1
#> 10 carcinoma_1 TRUE 1 5.05e-2 5.05e-2 1 1
#> 11 carcinoma_10 TRUE 1 1 e+0 1 e+0 1 1
#> 12 carcinoma_2 TRUE 1 1 e+0 1 e+0 1 1
#> 13 carcinoma_3 TRUE 1 1 e+0 1 e+0 1 1
#> 14 carcinoma_5 TRUE 1 1 e+0 1 e+0 1 1
#> 15 carcinoma_6 TRUE 1 1 e+0 1 e+0 1 1
#> 16 carcinoma_7 TRUE 1 1 e+0 1 e+0 1 1
#> 17 carcinoma_8 TRUE 1 1 e+0 1 e+0 1 1
#> 18 carcinoma_9_distal TRUE 1 1 e+0 1 e+0 1 1
#> 19 carcinoma_9_proxi… TRUE 1 1 e+0 1 e+0 1 1
#> # ℹ 2 more variables: converged <lgl>, penalty <dbl>
#>
#> =-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-
#> 2] Transfering orderings across patients
#> =-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-
#> [1] "w"
#>
#> =-=-=-=-=-=-=-=-=-=-=-=-=-=-=-
#> 1] Expectation Maximization
#> =-=-=-=-=-=-=-=-=-=-=-=-=-=-=-
#>
#> Number of Solutions
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Warning in sprintf("%4s", p, "|", sep = ""): 2 arguments not used by format
#> '%4s'
#> 1
#> Combinations of Transfer 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
#>
#> Initialization 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
#>
#> # 1 : E: OK M: 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |# A tibble: 19 × 9
#> patientID hasTrees numTrees maxScore minScore combInfTransf Solution
#> <chr> <lgl> <int> <dbl> <dbl> <int> <int>
#> 1 adenoma_1 TRUE 1 1 e+0 1 e+0 1 1
#> 2 adenoma_2 TRUE 1 1.13e-2 1.13e-2 1 1
#> 3 adenoma_3 TRUE 1 8.52e-5 8.52e-5 1 1
#> 4 adenoma_4 TRUE 1 2.55e-3 2.55e-3 1 1
#> 5 adenoma_5 TRUE 1 1 e+0 1 e+0 1 1
#> 6 adenoma_6 TRUE 1 1 e+0 1 e+0 1 1
#> 7 adenoma_7 TRUE 1 1 e+0 1 e+0 1 1
#> 8 adenoma_8 TRUE 1 1 e+0 1 e+0 1 1
#> 9 adenoma_9 TRUE 1 1 e+0 1 e+0 1 1
#> 10 carcinoma_1 TRUE 1 5.05e-2 5.05e-2 1 1
#> 11 carcinoma_10 TRUE 1 1 e+0 1 e+0 1 1
#> 12 carcinoma_2 TRUE 1 1 e+0 1 e+0 1 1
#> 13 carcinoma_3 TRUE 1 1 e+0 1 e+0 1 1
#> 14 carcinoma_5 TRUE 1 1 e+0 1 e+0 1 1
#> 15 carcinoma_6 TRUE 1 1 e+0 1 e+0 1 1
#> 16 carcinoma_7 TRUE 1 1 e+0 1 e+0 1 1
#> 17 carcinoma_8 TRUE 1 1 e+0 1 e+0 1 1
#> 18 carcinoma_9_distal TRUE 1 1 e+0 1 e+0 1 1
#> 19 carcinoma_9_proxi… TRUE 1 1 e+0 1 e+0 1 1
#> # ℹ 2 more variables: converged <lgl>, penalty <dbl>
#>
#> =-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-
#> 2] Transfering orderings across patients
#> =-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-
#>
#> =-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-
#> Selecting solution with minimal median penalty
#> =-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-
#> Solution #1 0.857142857142857
#> Solution #2 0.857142857142857
#> Solution #3 0.857142857142857
#> Best solution is # 1
#> REVOLVER Transfer Learning fit COMPLETED
#> Computing REVOLVER hierarchical clusters
CROSS_CRC_ADENOCARCINOMA_REVOLVER = revolver_cluster(
CROSS_CRC_ADENOCARCINOMA_REVOLVER,
split.method = 'cutreeHybrid',
min.group.size = 3)
#> [ REVOLVER Clustering - Colorectal adenocarcinomas (Cross et al, PMID 30177804) ]
#>
#> =-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-
#> Computing REVOLVER's evolutionary distance from the Information Transfer
#> =-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-
#>
#> Patients : N = 19 (171 comparisons)
#>
#>
#> =-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-
#> Computing Hierarchical Clustering from the distance
#> =-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-
#>
#> Clustering method ward
#> Split method cutreeHybrid
#> Minimum group size 3
#>
#>
#> Clusters : K = 3
#> Cluster size (n)
#> # A tibble: 3 × 2
#> cluster n
#> <chr> <int>
#> 1 C1 8
#> 2 C2 7
#> 3 C3 4
plot_clusters(CROSS_CRC_ADENOCARCINOMA_REVOLVER, cutoff_trajectories = 1, cutoff_drivers = 0)
#> Warning in rbind(deparse.level, ...): number of columns of result, 5, is not a
#> multiple of vector length 12 of arg 2
#> Warning in rbind(deparse.level, ...): number of columns of result, 5, is not a
#> multiple of vector length 12 of arg 2
#> Warning in rbind(deparse.level, ...): number of columns of result, 5, is not a
#> multiple of vector length 12 of arg 2
#> Warning in rbind(deparse.level, ...): number of columns of result, 5, is not a
#> multiple of vector length 12 of arg 2
#> Warning in rbind(deparse.level, ...): number of columns of result, 5, is not a
#> multiple of vector length 12 of arg 2
#> Warning in rbind(deparse.level, ...): number of columns of result, 5, is not a
#> multiple of vector length 12 of arg 2
#> Warning in rbind(deparse.level, ...): number of columns of result, 5, is not a
#> multiple of vector length 12 of arg 2
#> Warning in rbind(deparse.level, ...): number of columns of result, 5, is not a
#> multiple of vector length 12 of arg 2
#> Warning in rbind(deparse.level, ...): number of columns of result, 5, is not a
#> multiple of vector length 12 of arg 2
#> Warning in rbind(deparse.level, ...): number of columns of result, 5, is not a
#> multiple of vector length 12 of arg 2
#> Warning in rbind(deparse.level, ...): number of columns of result, 5, is not a
#> multiple of vector length 12 of arg 2
#> Warning in rbind(deparse.level, ...): number of columns of result, 5, is not a
#> multiple of vector length 12 of arg 2
#> Warning in rbind(deparse.level, ...): number of columns of result, 5, is not a
#> multiple of vector length 12 of arg 2
#> Warning in rbind(deparse.level, ...): number of columns of result, 5, is not a
#> multiple of vector length 12 of arg 2
#> Warning in rbind(deparse.level, ...): number of columns of result, 5, is not a
#> multiple of vector length 12 of arg 2
#> Warning in rbind(deparse.level, ...): number of columns of result, 5, is not a
#> multiple of vector length 12 of arg 2
#> Warning: Using `size` aesthetic for lines was deprecated in ggplot2 3.4.0.
#> ℹ Please use `linewidth` instead.
#> ℹ The deprecated feature was likely used in the revolver package.
#> Please report the issue at <https://github.com/caravagnalab/revolver/issues>.
#> This warning is displayed once per session.
#> Call `lifecycle::last_lifecycle_warnings()` to see where this warning was
#> generated.
#> Warning: No shared levels found between `names(values)` of the manual scale and the
#> data's colour values.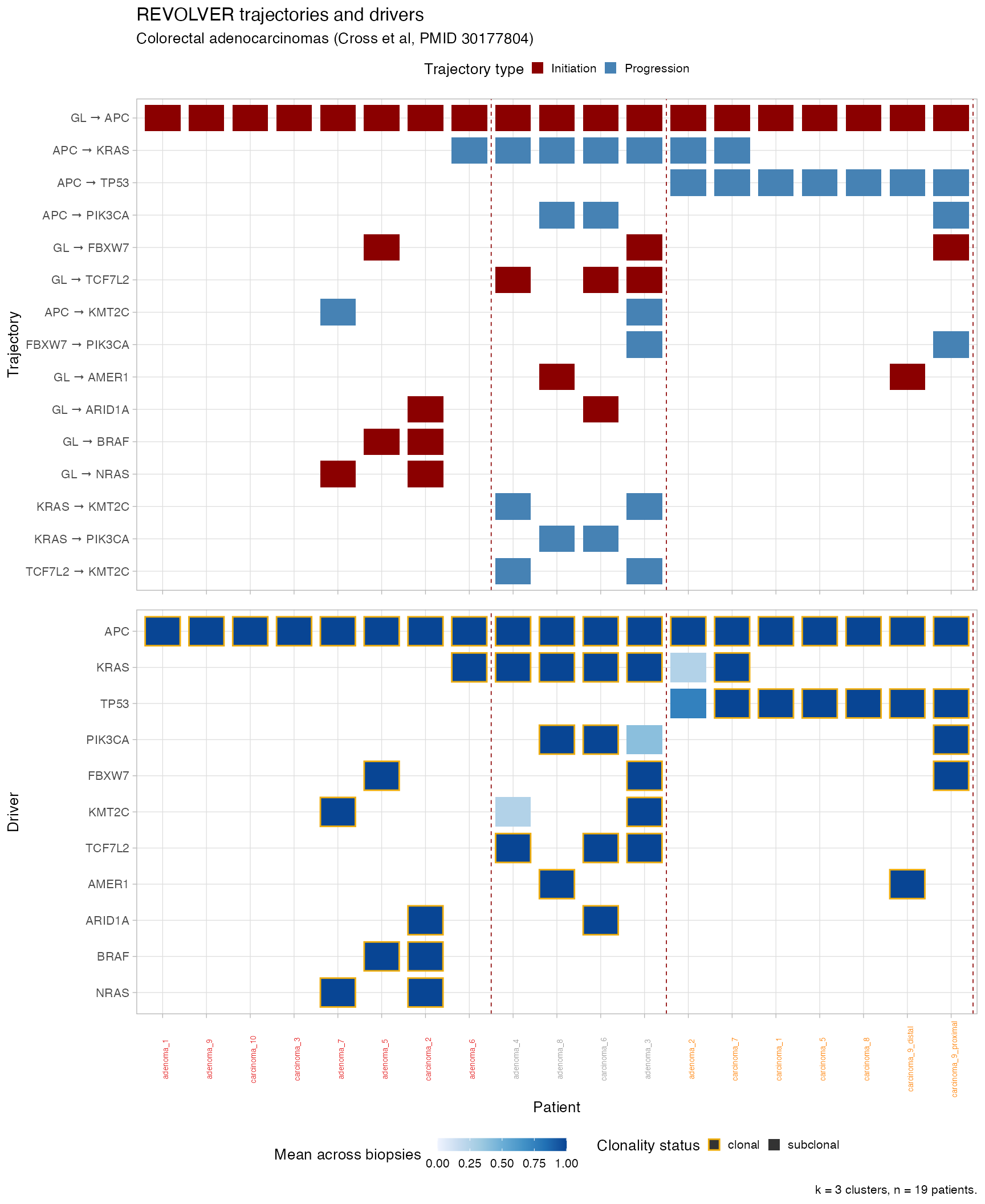
plot_drivers_graph(CROSS_CRC_ADENOCARCINOMA_REVOLVER)
#>
#> =-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-
#> Enrichment test for incoming edges
#> =-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-
#> # A tibble: 3 × 15
#> estimate p.value conf.low conf.high method alternative from to POS_POS
#> <dbl> <dbl> <dbl> <dbl> <chr> <chr> <chr> <chr> <int>
#> 1 Inf 0.000000191 10.1 Inf Fishe… greater GL APC 19
#> 2 Inf 0.000102 5.63 Inf Fishe… greater APC KRAS 7
#> 3 Inf 0.000102 5.63 Inf Fishe… greater APC TP53 7
#> # ℹ 6 more variables: POS_NEG <int>, NEG_POS <int>, NEG_NEG <int>,
#> # alpha_level <dbl>, N <int>, psign <lgl>
#> Warning: Removed 1 row containing missing values or values outside the scale range
#> (`geom_point()`).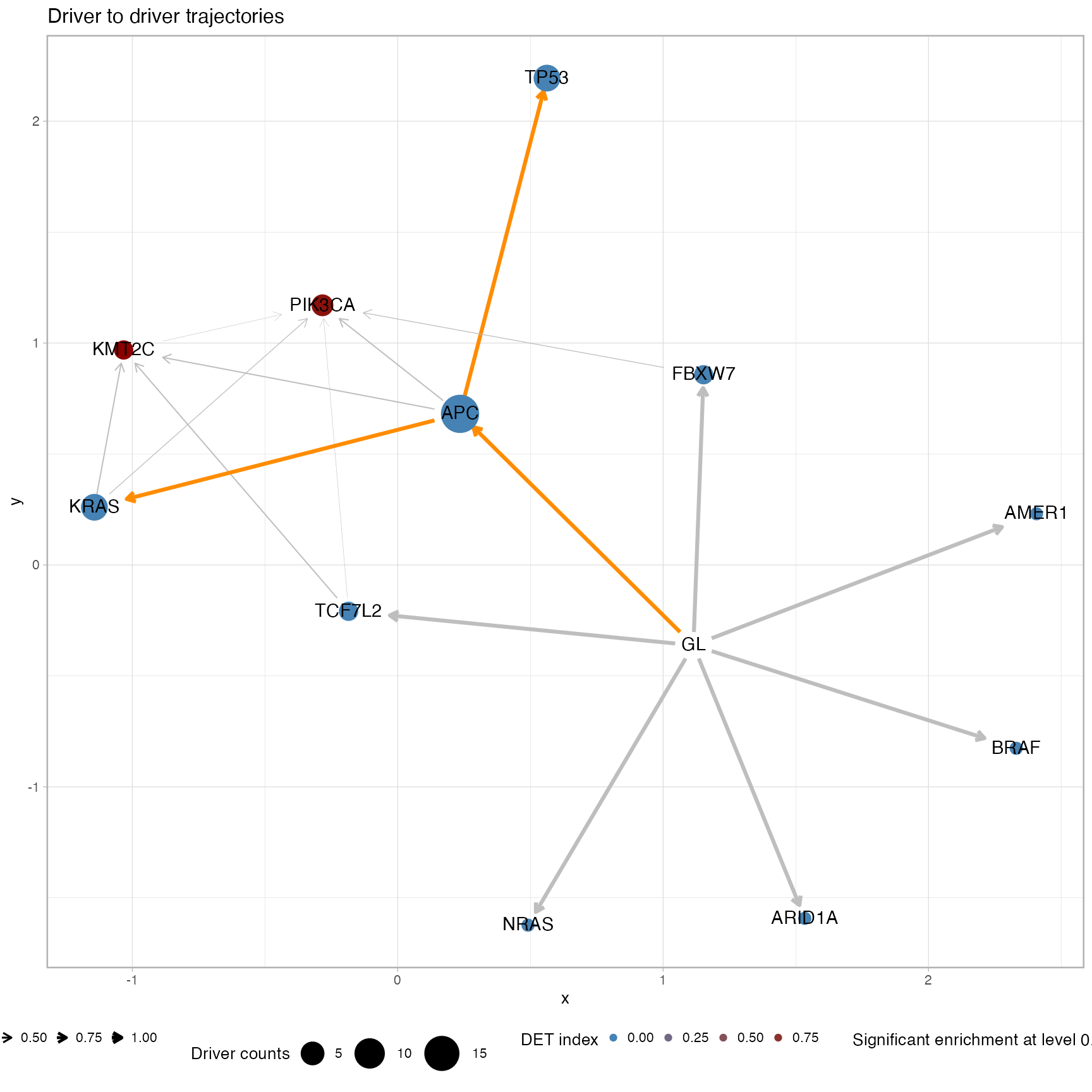
revolver::plot_dendrogram(CROSS_CRC_ADENOCARCINOMA_REVOLVER)
#> Warning: No shared levels found between `names(values)` of the manual scale and the
#> data's colour values.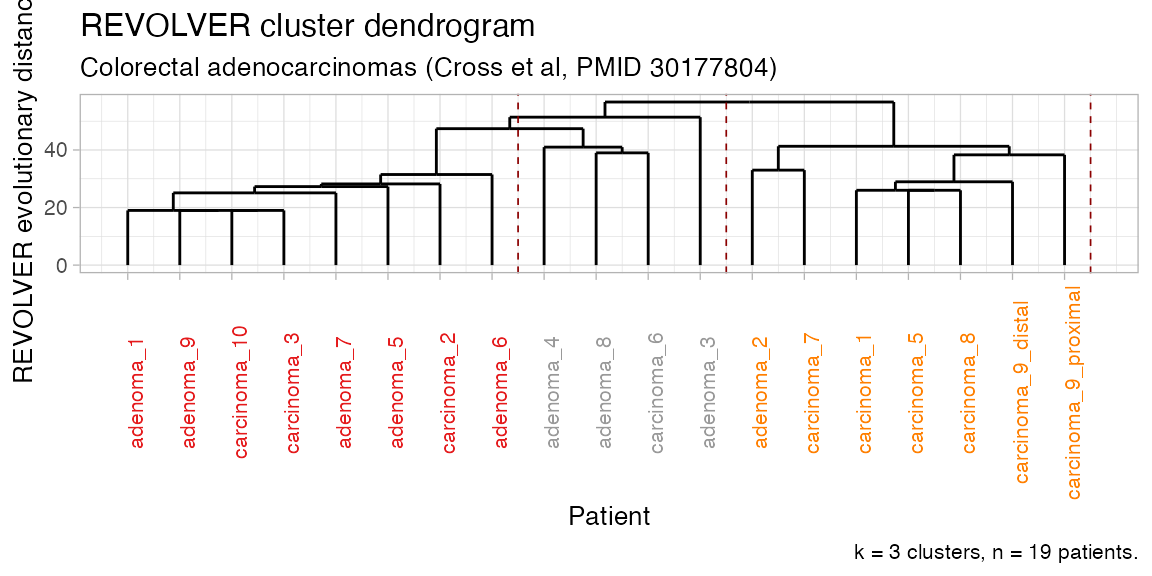
plot_DET_index(CROSS_CRC_ADENOCARCINOMA_REVOLVER)
#> # A tibble: 11 × 4
#> driver diversity N DET_index
#> <chr> <dbl> <int> <dbl>
#> 1 AMER1 0 1 0
#> 2 APC 0 1 0
#> 3 ARID1A 0 1 0
#> 4 BRAF 0 1 0
#> 5 FBXW7 0 1 0
#> 6 KRAS 0 1 0
#> 7 NRAS 0 1 0
#> 8 TCF7L2 0 1 0
#> 9 TP53 0 1 0
#> 10 PIK3CA 1.52 5 0.946
#> 11 KMT2C 1.10 3 1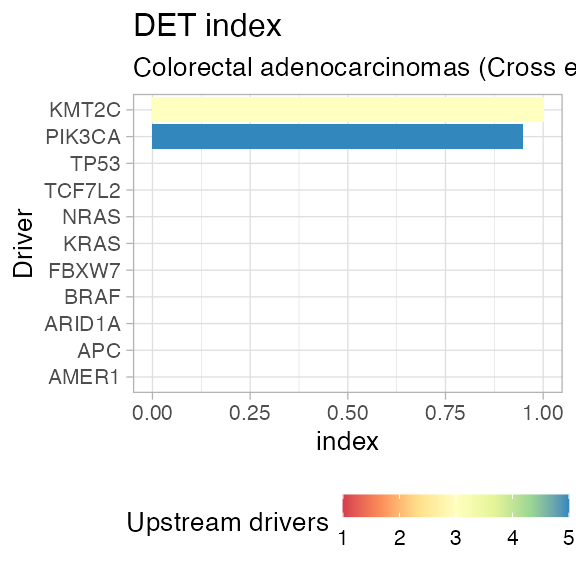
plot(CROSS_CRC_ADENOCARCINOMA_REVOLVER)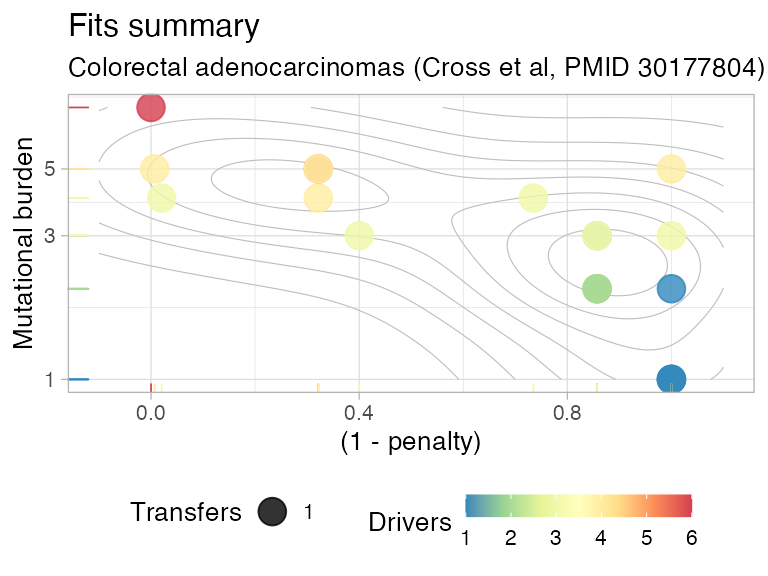
plot_drivers_clonality(CROSS_CRC_ADENOCARCINOMA_REVOLVER)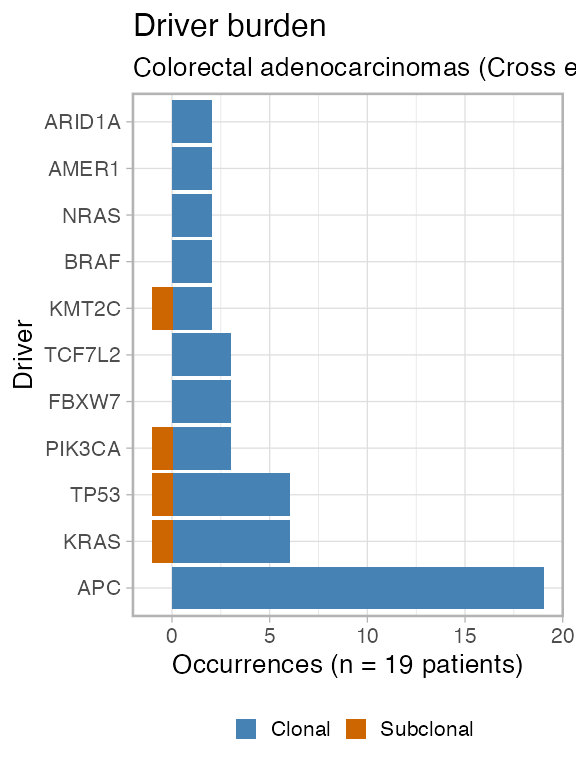
plot_drivers_occurrence(CROSS_CRC_ADENOCARCINOMA_REVOLVER)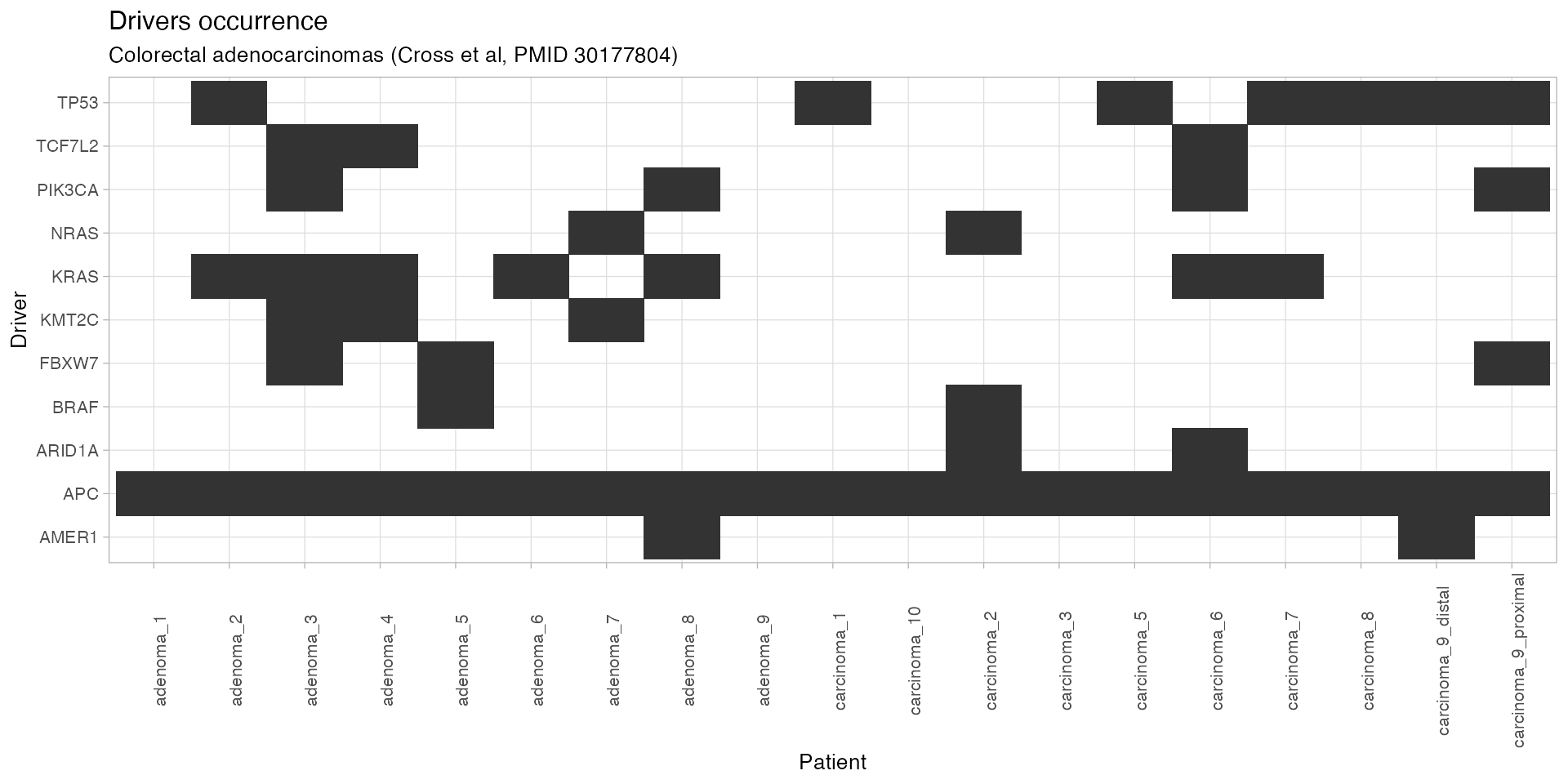
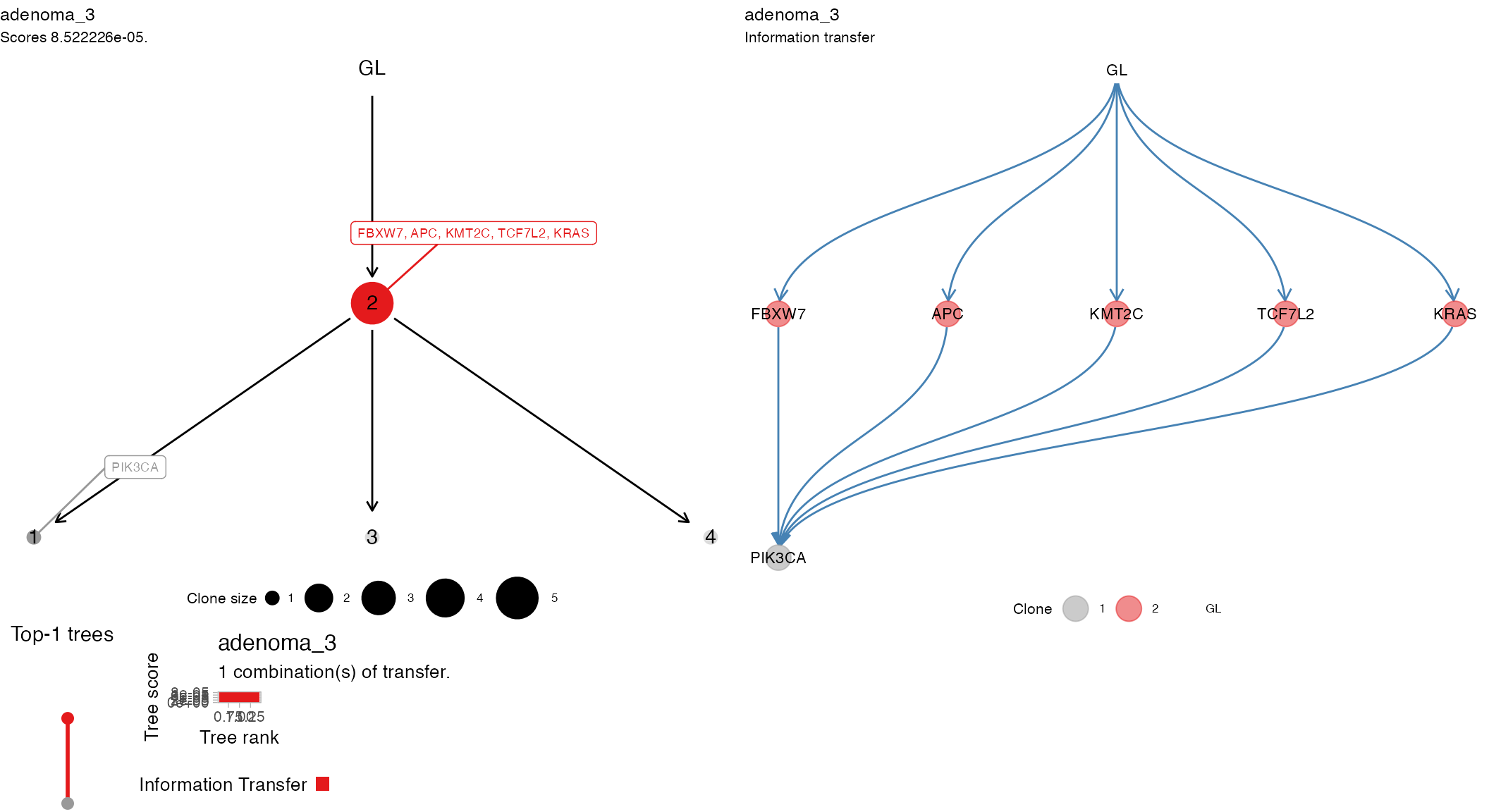
plot_patient_trees(CROSS_CRC_ADENOCARCINOMA_REVOLVER, CROSS_CRC_ADENOCARCINOMA_REVOLVER$patients[3])
#> Warning: The `<scale>` argument of `guides()` cannot be `FALSE`. Use "none" instead as
#> of ggplot2 3.3.4.
#> ℹ The deprecated feature was likely used in the mtree package.
#> Please report the issue to the authors.
#> This warning is displayed once per session.
#> Call `lifecycle::last_lifecycle_warnings()` to see where this warning was
#> generated.