Input formats and creation of a REVOLVER cohort
Source:vignettes/Input_formats.Rmd
Input_formats.RmdInput format for REVOLVER
Data from a cohort of patients can be represented as a dataframe with at least 7 columns, where every row represents one genomic alteration annotated for the analysis.
| Field | Type | Description |
|---|---|---|
Misc |
string | Customary annotation which is never used to analyse the data, but that might be good to carry around |
patientID |
string | A patient identifier |
variantID |
string | An alteration identifier |
cluster |
string | Group ID (eg., a clone, with CCF data) |
is.driver |
logical | TRUE if the alteration is a driver to correlate when repeated trajectores are extracted |
is.clonal |
logical | TRUE if the alteration belongs to the group of clonal (truncal) events; there should only one such clonal group per patient |
CCF |
string | A parsable format for storage of input CCFs or binary data (see below) |
All identifiers cannot contain spaces or dash/ hyphen
(-) symbols, and must be unique:
the
patientIDmust be unique across patients;the
variantIDfor driver events (those withis.driver = TRUE) must be shared across all patients that have the same driver event, and cannot appear multiple times in the data of a single patient. Notice that you should not annotate an alteration as driver if it is not found in multiple patients.the
variantIDfor non-driver events (those withis.driver = FALSE) is in practice never used and therefore can be set freely.
Notice that the input dataframe has the same structure for both CCF and binary input data.
Supported alterations
The scope of supported alterations is broad, and include any SNV, larger chromosomal re-arrangment or other covariate that can be encoded in CCF or binary format.
The variantID field of driver alterations
(is.driver=TRUE) will be matched to correlate trajectories
in multiple patients, and can be you whatever you find more suitable for
your analysis, for instance:
a Hugo_Symbol (
BRAF)a name for a well-known SNV (
BRAF_600E)a reference to some cytoband (
3q26.32)your custom annotation (
MyFavoritePathway).
Alterations are also associated to groups (via cluster),
which constitute the nodes of the computed trees. A group can have 0
drivers annotated, but every patient should have at least one driver to
be analyzed with REVOLVER.
See also the Frequently Asked Questions if you are interested in modelling parallel evolution.
Input values for CCF or binary datasets
Field CCF represents data from both types of supported
datasets:
real-valued CCF values (in [0, 1]),
or input binary values (either 0 or 1).
Since patients can have different number of samples/ regions
associated, CCF is a general string.
The format that we propose is simple, and easy to parse it:
# Example string that reports a CCF value 0.86 in region R1, 1 in R2 etc.
R1:0.86;R2:1;R3:1
# Similarly, in binary presence/ absence format ...
R1:0;R2:1;R3:1If you use this format, REVOLVER provides you a parsing
function that you can pass to the cohort
construction function
Example data
An example cohort is released in the evoverse.datasets
R package,
which also provides a REVOLVER objectwith the final
analysis of the input cohort.
# Load the package and runs available_data() to print what is available
library(evoverse.datasets)
# Load an example dataset - the TRACERx cohort
data("TRACERx_NEJM_2017", package = "evoverse.datasets")
# This is a dataset that follows the specifications given above
print(TRACERx_NEJM_2017)
#> # A tibble: 65,421 × 7
#> Misc patientID variantID cluster is.driver is.clonal CCF
#> <chr> <chr> <chr> <chr> <lgl> <lgl> <chr>
#> 1 CRUK0001:7:47971467:C CRUK0001 PKD1L1 1 FALSE FALSE R1:0…
#> 2 CRUK0001:21:47754753:C CRUK0001 PCNT 1 FALSE FALSE R1:0…
#> 3 CRUK0001:22:37331483:G CRUK0001 CSF2RB 1 FALSE FALSE R1:0…
#> 4 CRUK0001:10:102824636:G CRUK0001 KAZALD1 1 FALSE FALSE R1:0…
#> 5 CRUK0001:7:81978881:C CRUK0001 CACNA2D1 1 FALSE FALSE R1:0…
#> 6 CRUK0001:1:201843493:G CRUK0001 IPO9 1 FALSE FALSE R1:0…
#> 7 CRUK0001:3:167402119:G CRUK0001 PDCD10 1 FALSE FALSE R1:0…
#> 8 CRUK0001:7:134650062:G CRUK0001 CALD1 1 FALSE FALSE R1:0…
#> 9 CRUK0001:17:76989965:C CRUK0001 CANT1 1 FALSE FALSE R1:0…
#> 10 CRUK0001:16:1662386:G CRUK0001 IFT140 1 FALSE FALSE R1:0…
#> # ℹ 65,411 more rowsCohort construction
Once you have a dataframe/ tibble in the required format, you can use a cohort construction function.
In this case, to make it faster we only use 3 patients from the released cohort.
library(dplyr)
#>
#> Attaching package: 'dplyr'
#> The following objects are masked from 'package:stats':
#>
#> filter, lag
#> The following objects are masked from 'package:base':
#>
#> intersect, setdiff, setequal, union
require(revolver)
#> Loading required package: revolver
#> Warning: replacing previous import 'cli::num_ansi_colors' by
#> 'crayon::num_ansi_colors' when loading 'easypar'
#> Loading revolver, 'Repeated Evolution in Cancer'. Support : https://caravagn.github.io/revolver/
subset_data = TRACERx_NEJM_2017 %>%
filter(patientID %in% c("CRUK0001", "CRUK0002", "CRUK0003"))
# Create a cohort
my_cohort = revolver_cohort(
subset_data,
CCF_parser = CCF_parser,
ONLY.DRIVER = FALSE,
MIN.CLUSTER.SIZE = 0,
annotation = "My small REVOLVER dataset"
)
#> [ REVOLVER ~ Cohort constructor ]
#>
#> ℹ Using only driver mutations.
#> ℹ Rejecting clusters with less than 0 mutations.
#>
#> ── REVOLVER input data ─────────────────────────────────────────────────────────
#>
#> # A tibble: 2,710 × 9
#> Misc patientID variantID cluster is.driver is.clonal CCF id
#> <chr> <chr> <chr> <chr> <lgl> <lgl> <chr> <chr>
#> 1 CRUK0001:7:47971… CRUK0001 PKD1L1 1 FALSE FALSE R1:0… __mu…
#> 2 CRUK0001:21:4775… CRUK0001 PCNT 1 FALSE FALSE R1:0… __mu…
#> 3 CRUK0001:22:3733… CRUK0001 CSF2RB 1 FALSE FALSE R1:0… __mu…
#> 4 CRUK0001:10:1028… CRUK0001 KAZALD1 1 FALSE FALSE R1:0… __mu…
#> 5 CRUK0001:7:81978… CRUK0001 CACNA2D1 1 FALSE FALSE R1:0… __mu…
#> 6 CRUK0001:1:20184… CRUK0001 IPO9 1 FALSE FALSE R1:0… __mu…
#> 7 CRUK0001:3:16740… CRUK0001 PDCD10 1 FALSE FALSE R1:0… __mu…
#> 8 CRUK0001:7:13465… CRUK0001 CALD1 1 FALSE FALSE R1:0… __mu…
#> 9 CRUK0001:17:7698… CRUK0001 CANT1 1 FALSE FALSE R1:0… __mu…
#> 10 CRUK0001:16:1662… CRUK0001 IFT140 1 FALSE FALSE R1:0… __mu…
#> # ℹ 2,700 more rows
#> # ℹ 1 more variable: cluster_size <int>
#>
#> ── Preprocessing data (this may take some time)
#>
#> ...
#>
#> ── Extracting clones table ─────────────────────────────────────────────────────
#> → CRUK0001 : 2100 entries, 11 clone(s).
#> → CRUK0002 : 280 entries, 7 clone(s).
#> → CRUK0003 : 330 entries, 7 clone(s).
# This is now a REVOLVER cohort with S3 methods available
print(my_cohort)
#> [ REVOLVER - Repeated Evolution in Cancer ]
#>
#> Dataset : My small REVOLVER dataset
#> Cohort : 3 patients, 2710 variants and 16 driver events.
#>
#> Trees per patient : NO
#> Fit via TL : NO
#> REVOLVER clustering : NO
#> Jackknife statistics : NO
#>
#> For summary statistics see `?Stats_*(x)` with * = {cohort, drivers, trees, fits, clusters, ...}
#>
#> ┌──────────────────────────────────────────────────────────────────────┐
#> │ │
#> │ WARNING - Driver variantIDs occuring only once could be removed. │
#> │ │
#> └──────────────────────────────────────────────────────────────────────┘
#> # A tibble: 14 × 7
#> variantID numClonal p_clonal numSubclonal p_subclonal N_tot p_tot
#> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
#> 1 TP53 1 0.333 0 0 1 0.333
#> 2 MGA 1 0.333 0 0 1 0.333
#> 3 WRN 1 0.333 0 0 1 0.333
#> 4 MET 1 0.333 0 0 1 0.333
#> 5 TERT 1 0.333 0 0 1 0.333
#> 6 PIK3CA 1 0.333 0 0 1 0.333
#> 7 CDKN2A 1 0.333 0 0 1 0.333
#> 8 ARHGAP35 0 0 1 0.333 1 0.333
#> 9 PASK 0 0 1 0.333 1 0.333
#> 10 RB1 0 0 1 0.333 1 0.333
#> 11 IKZF1 0 0 1 0.333 1 0.333
#> 12 KRAS 0 0 1 0.333 1 0.333
#> 13 EP300 0 0 1 0.333 1 0.333
#> 14 CTNNB1 0 0 1 0.333 1 0.333
plot(my_cohort)
#> Warning: Using `size` aesthetic for lines was deprecated in ggplot2 3.4.0.
#> ℹ Please use `linewidth` instead.
#> ℹ The deprecated feature was likely used in the revolver package.
#> Please report the issue at <https://github.com/caravagnalab/revolver/issues>.
#> This warning is displayed once per session.
#> Call `lifecycle::last_lifecycle_warnings()` to see where this warning was
#> generated.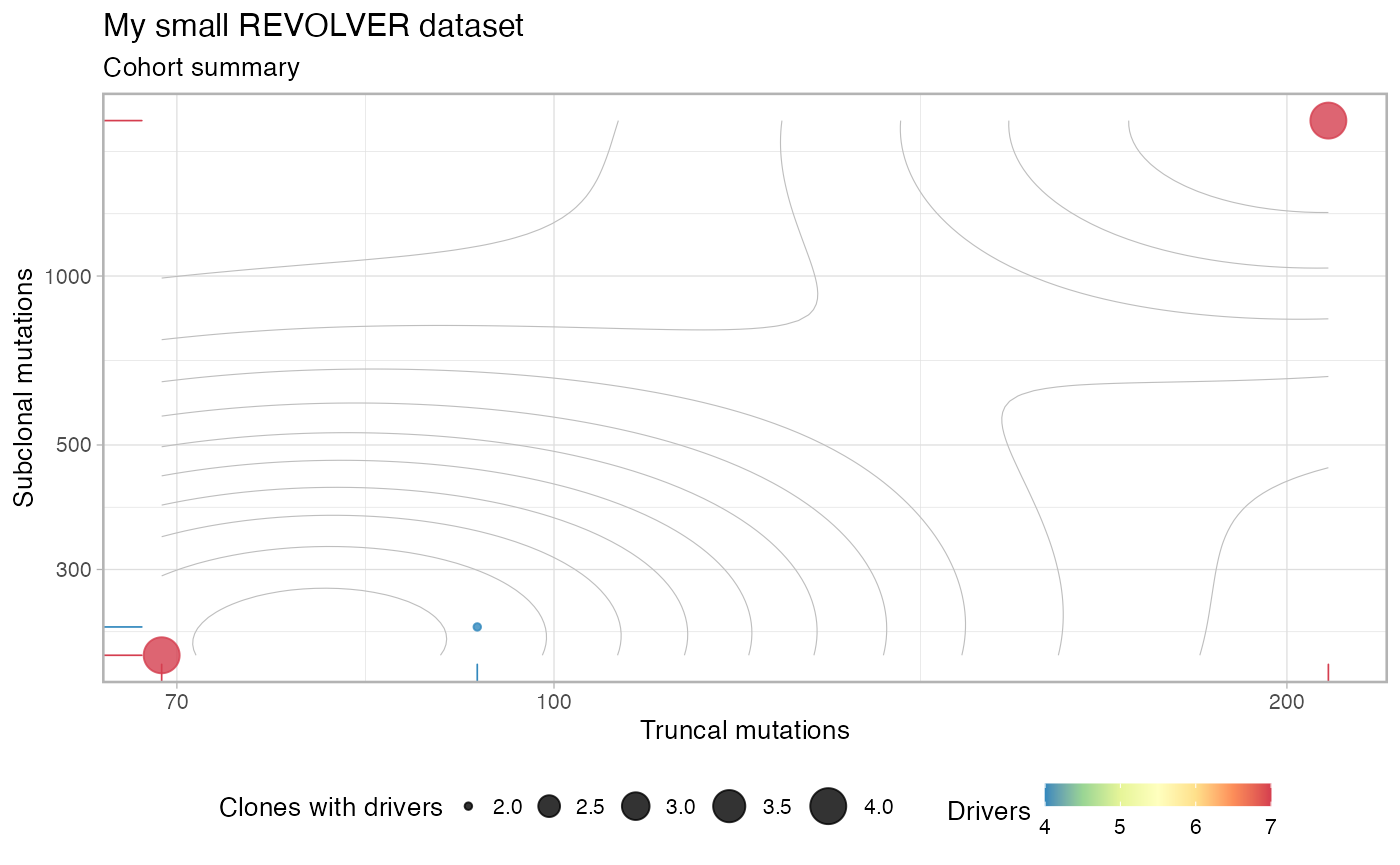
The cohort is now ready to be inspected and analysed (note that since we only used 3 patients a warning is raised to mention that the cohort contains drivers annotated which are not really recurrent and that, therefore, should be removed; if you construct the cohort object with the full set of 99 patients that warning will vanish).
# Load a full cohort - the TRACERx cohort
data("TRACERx_NEJM_2017_REVOLVER", package = "evoverse.datasets")
# We can use S3 object functions to retrieve simple information about the cohort.
# The `print` functions runs also the `revolver_check_cohort` function which
# tells us that some patient have only 1 clone with drivers, and therefore they
# can just be expanded.
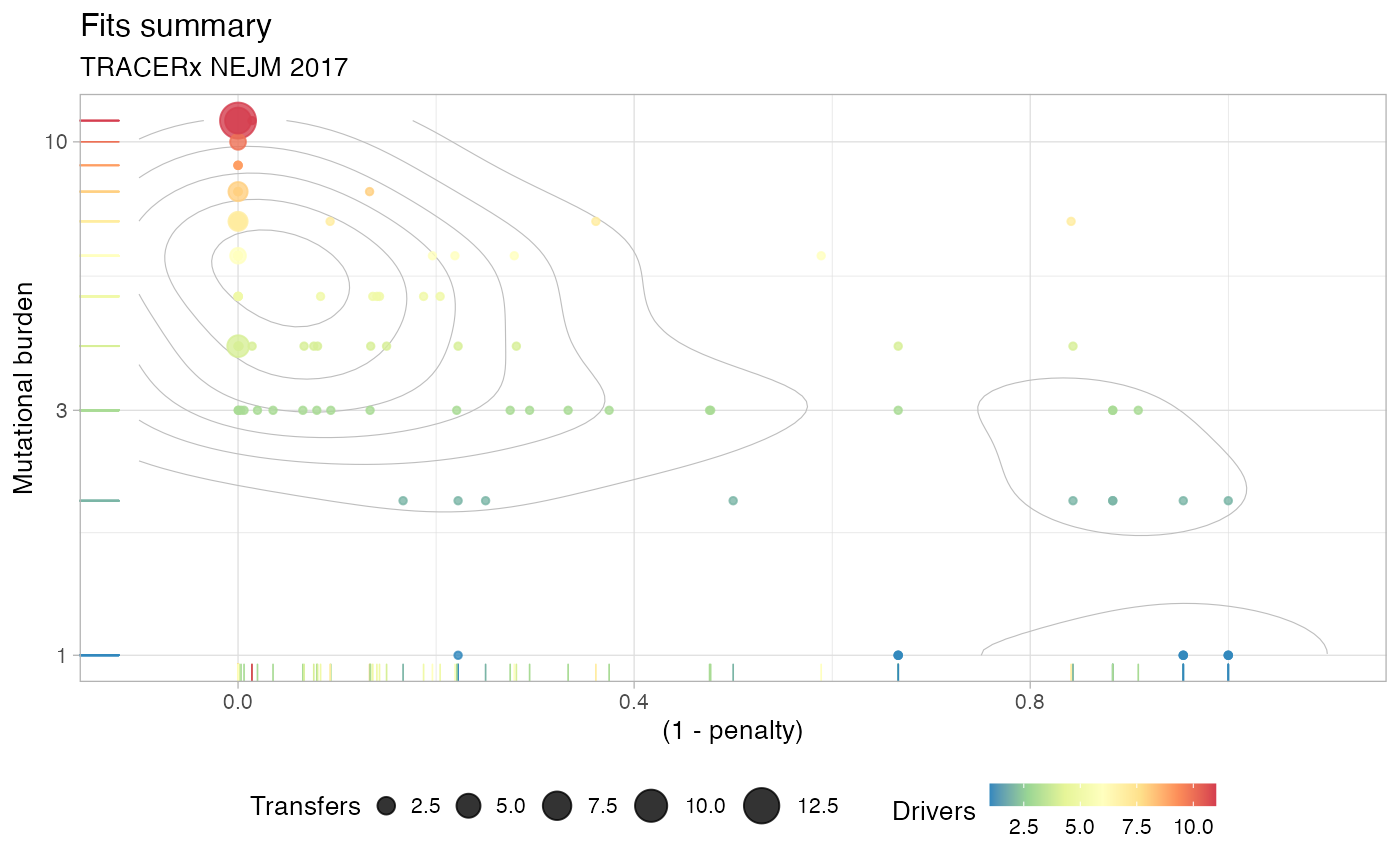
print(TRACERx_NEJM_2017_REVOLVER)
#> [ REVOLVER - Repeated Evolution in Cancer ]
#>
#> Dataset : TRACERx NEJM 2017
#> Cohort : 99 patients, 450 variants and 79 driver events.
#>
#> Trees per patient : YES
#> Fit via TL : YES
#> REVOLVER clustering : YES
#> Jackknife statistics : YES
#>
#> For summary statistics see `?Stats_*(x)` with * = {cohort, drivers, trees, fits, clusters, ...}
#>
#> ┌───────────────────────────────────────────────────────────────────────────────────────────┐
#> │ │
#> │ WARNING - Some patients have only one clone with drivers; they will just be expanded. │
#> │ │
#> └───────────────────────────────────────────────────────────────────────────────────────────┘
#> # A tibble: 54 × 7
#> patientID numBiopsies numMutations numDriverMutations numClonesWithDriver
#> <chr> <int> <int> <int> <int>
#> 1 CRUK0007 2 3 3 1
#> 2 CRUK0010 2 3 3 1
#> 3 CRUK0012 2 1 1 1
#> 4 CRUK0018 4 4 4 1
#> 5 CRUK0019 2 1 1 1
#> 6 CRUK0021 2 4 4 1
#> 7 CRUK0025 3 3 3 1
#> 8 CRUK0026 2 4 4 1
#> 9 CRUK0028 2 2 2 1
#> 10 CRUK0029 6 4 4 1
#> # ℹ 44 more rows
#> # ℹ 2 more variables: numTruncalMutations <int>, numSubclonalMutations <int>
# Plot
plot(TRACERx_NEJM_2017_REVOLVER)