library(CNAqc)
#> ✔ Loading CNAqc, 'Copy Number Alteration quality check'. Support : <https://caravagn.github.io/CNAqc/>Fragmentation of individual arms
The fragmentation of a chromosome arm is assessed with a statistical test based on counting the size of the copy number segments mapping to the arm. This analysis works only at the level of clonal segments
We work with the template dataset.
#>
#> 2:2 [n = 7478, L = 1483 Mb] ■■■■■■■■■■■■■■■■■■■■■■■■■■■ { CTCF }
#> 4:2 [n = 1893, L = 331 Mb] ■■■■■■■
#> 3:2 [n = 1625, L = 357 Mb] ■■■■■■
#> 2:1 [n = 1563, L = 420 Mb] ■■■■■■ { TTN }
#> 3:0 [n = 312, L = 137 Mb] ■
#> 2:0 [n = 81, L = 39 Mb] { TP53 }
#> 16:2 [n = 4, L = 0 Mb]
#> 25:2 [n = 2, L = 1 Mb]
#> 3:1 [n = 2, L = 1 Mb]
#> 106:1 [n = 1, L = 0 Mb]
#> chr from to ref alt DP NV VAF driver_label is_driver
#> chr2 179431633 179431634 C T 117 77 0.6581197 TTN TRUE
#> chr16 67646006 67646007 C T 120 54 0.4500000 CTCF TRUE
#> chr17 7577106 7577107 G C 84 78 0.9285714 TP53 TRUE
# A histogram of segments' lenght
plot_segment_size_distribution(x)
#> Warning: Using `size` aesthetic for lines was deprecated in ggplot2 3.4.0.
#> ℹ Please use `linewidth` instead.
#> ℹ The deprecated feature was likely used in the CNAqc package.
#> Please report the issue at <https://github.com/caravagnalab/CNAqc/issues>.
#> This warning is displayed once every 8 hours.
#> Call `lifecycle::last_lifecycle_warnings()` to see where this warning was
#> generated.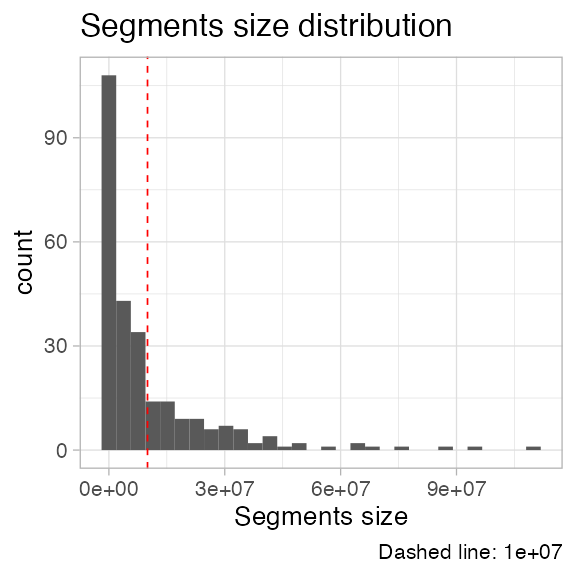
CNAqc counts, for every arm with lenght
nucleotides:
- , the number of mapped CNA segments shorter than of ;
- , the number of mapped CNA segments longer than sof .
A one-sided Binomial test is used to compute a p-value for the null hypothesis of seeing observations in trials, assuming a Binomial success probability . represents a model where each segment length is equally likely (uniform distribution).
In this way the test accounts for the difference in lenghts of the chromsome arms; a p-value per arm is reported and adjusted for multiple hyoptheses (Bonferroni).
# Test with default parameters (small segments: < 20% of chromosome arm)
x = detect_arm_overfragmentation(x)
#> ℹ One-tailed Binomial test: 8 tests, alpha 0.01. Short segments: 0.2% of the reference arm.
#> ✔ chr7p, p = 1.7179869184e-24 ~ 34 segments, 34 short.
#> ✔ chr1p, p = 1.627389952e-15 ~ 24 segments, 23 short.
#> ✔ chr1q, p = 4.34176000000001e-08 ~ 13 segments, 12 short.
#> ✔ chr11q, p = 1.0657792e-06 ~ 13 segments, 11 short.
#> ✔ chr12q, p = 2.00704e-07 ~ 12 segments, 11 short.
#> ✔ chr3q, p = 4.52608e-06 ~ 12 segments, 10 short.
#> ✔ chr7q, p = 4.52608e-06 ~ 12 segments, 10 short.
#> ✔ chr8p, p = 9.21599999999998e-07 ~ 11 segments, 10 short.
#> ℹ 8 significantly overfragmented chromosome arms (alpha level 0.01).
print(x)
#> ── [ CNAqc ] MySample 12963 mutations in 267 segments (267 clonal, 0 subclonal).
#>
#> ── Clonal CNAs
#>
#> 2:2 [n = 7478, L = 1483 Mb] ■■■■■■■■■■■■■■■■■■■■■■■■■■■ { CTCF }
#> 4:2 [n = 1893, L = 331 Mb] ■■■■■■■
#> 3:2 [n = 1625, L = 357 Mb] ■■■■■■
#> 2:1 [n = 1563, L = 420 Mb] ■■■■■■ { TTN }
#> 3:0 [n = 312, L = 137 Mb] ■
#> 2:0 [n = 81, L = 39 Mb] { TP53 }
#> 16:2 [n = 4, L = 0 Mb]
#> 25:2 [n = 2, L = 1 Mb]
#> 3:1 [n = 2, L = 1 Mb]
#> 106:1 [n = 1, L = 0 Mb]
#> ℹ Sample Purity: 89% ~ Ploidy: 4.
#> ℹ There are 3 annotated driver(s) mapped to clonal CNAs.
#> chr from to ref alt DP NV VAF driver_label is_driver
#> chr2 179431633 179431634 C T 117 77 0.6581197 TTN TRUE
#> chr16 67646006 67646007 C T 120 54 0.4500000 CTCF TRUE
#> chr17 7577106 7577107 G C 84 78 0.9285714 TP53 TRUE
#> ✔ Arm-level fragmentation analysis: 8 segments overfragmented.You can produce a arm-level report for the fragmentation test, with:
- a scatter of the counts per arm, with scaled the p-values;
- a jump statistics per arm, .
is the sum of the variation in total copy number profiles, evaluated among each pair of contiguous segments.
Significantly overfragmented arms with high have a “scattered” copy number profile. Those with low are more uniform, as they show little no copy number change, and can be possibly smoothed (see below).
plot_arm_fragmentation(x, zoom = 0)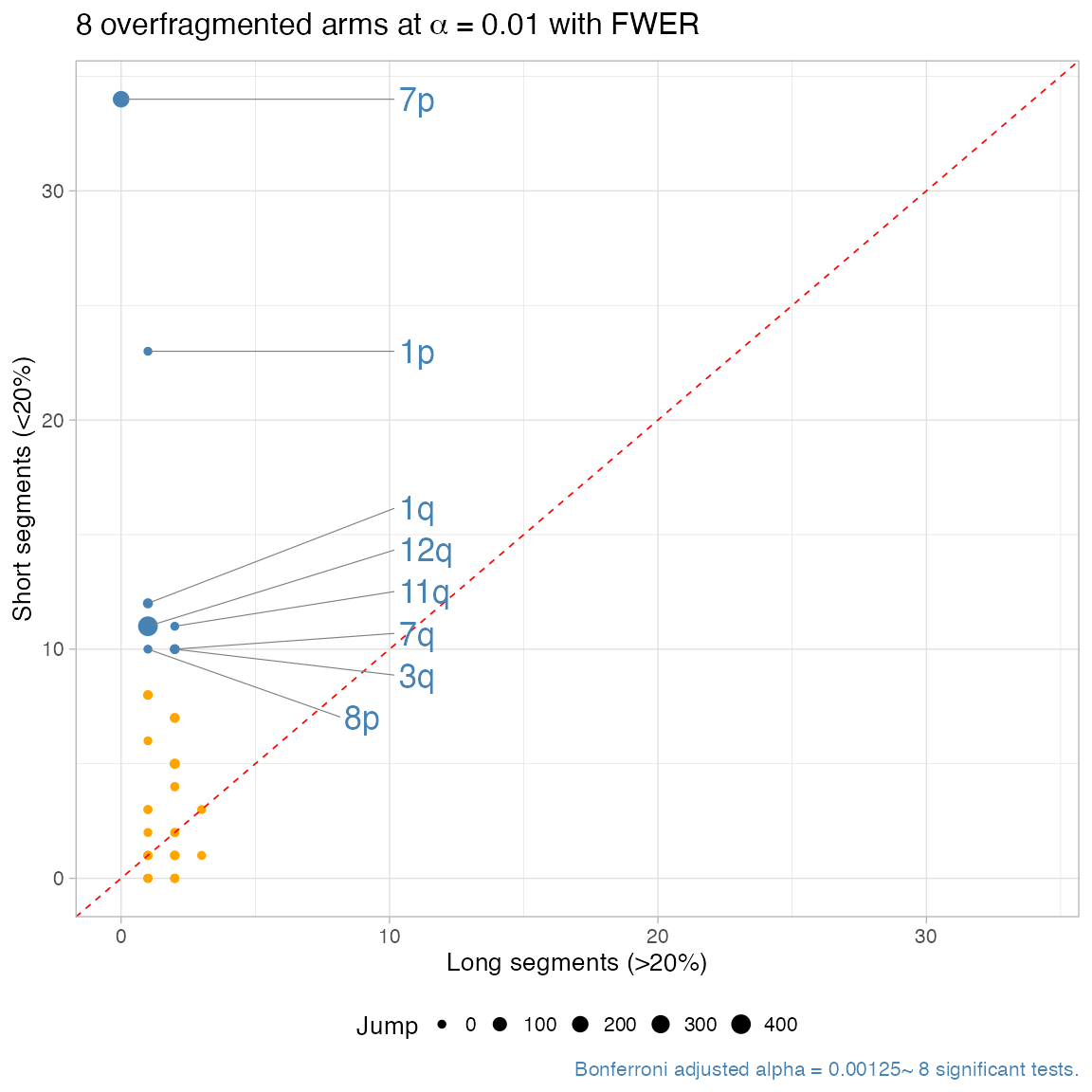
Once available, these results appear in any call to
plot_segments as annotated purple squares sorrounding the
arms.
# Default plot has now segments
plot_segments(x)
#> Scale for fill is already present.
#> Adding another scale for fill, which will replace the existing scale.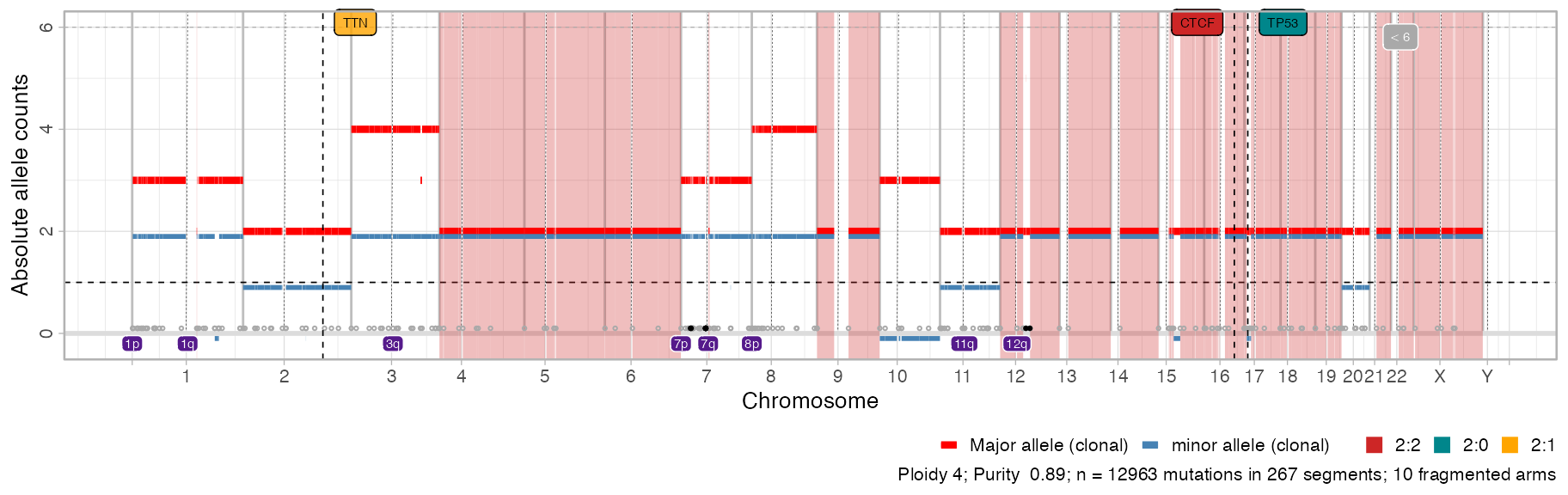
Smoothing is a good way to start cleaning up the fragmented sets of arms.
# Smooth with default parameters
x = smooth_segments(x)
#> → chr1 37 -6 @
#> → chr10 8 -3 @
#> → chr11 22 -3 @
#> → chr12 13 -11 @
#> → chr14 2 -1 @
#> → chr15 9 -3 @
#> → chr16 10 -3 @
#> → chr17 10 -6 @
#> → chr18 8 -2 @
#> → chr19 5 -2 @
#> → chr2 18 -5 @
#> → chr20 9 -2 @
#> → chr21 2 -1 @
#> → chr22 3 -3 @
#> → chr3 19 -4 @
#> → chr4 8 -2 @
#> → chr5 6 -3 @
#> → chr6 4 -2 @
#> → chr7 46 -17 @
#> → chr8 18 -3 @
#> → chr9 3 -2 @
#> → chrX 6 -2 @
#> ✔ Smoothed from 267 to 87 segments with 1e+06 gap (bases).
#> ℹ Creating a new CNAqc object. The old object will be retained in the $before_smoothing field.
#>
#> ── CNAqc - CNA Quality Check ───────────────────────────────────────────────────
#> ℹ Using reference genome coordinates for: hg19.
#> ✔ Found annotated driver mutations: TTN, CTCF, and TP53.
#> ✔ Fortified calls for 12963 somatic mutations: 12963 SNVs (100%) and 0 indels.
#> ✔ Fortified CNAs for 87 segments: 87 clonal and 0 subclonal.
#> Warning in map_mutations_to_clonal_segments(mutations, cna_clonal): [CNAqc] a
#> karyotype column is present in CNA calls, and will be overwritten
#> ✔ 12963 mutations mapped to clonal CNAs.
# Re-compute the fragmentation
x = detect_arm_overfragmentation(x)
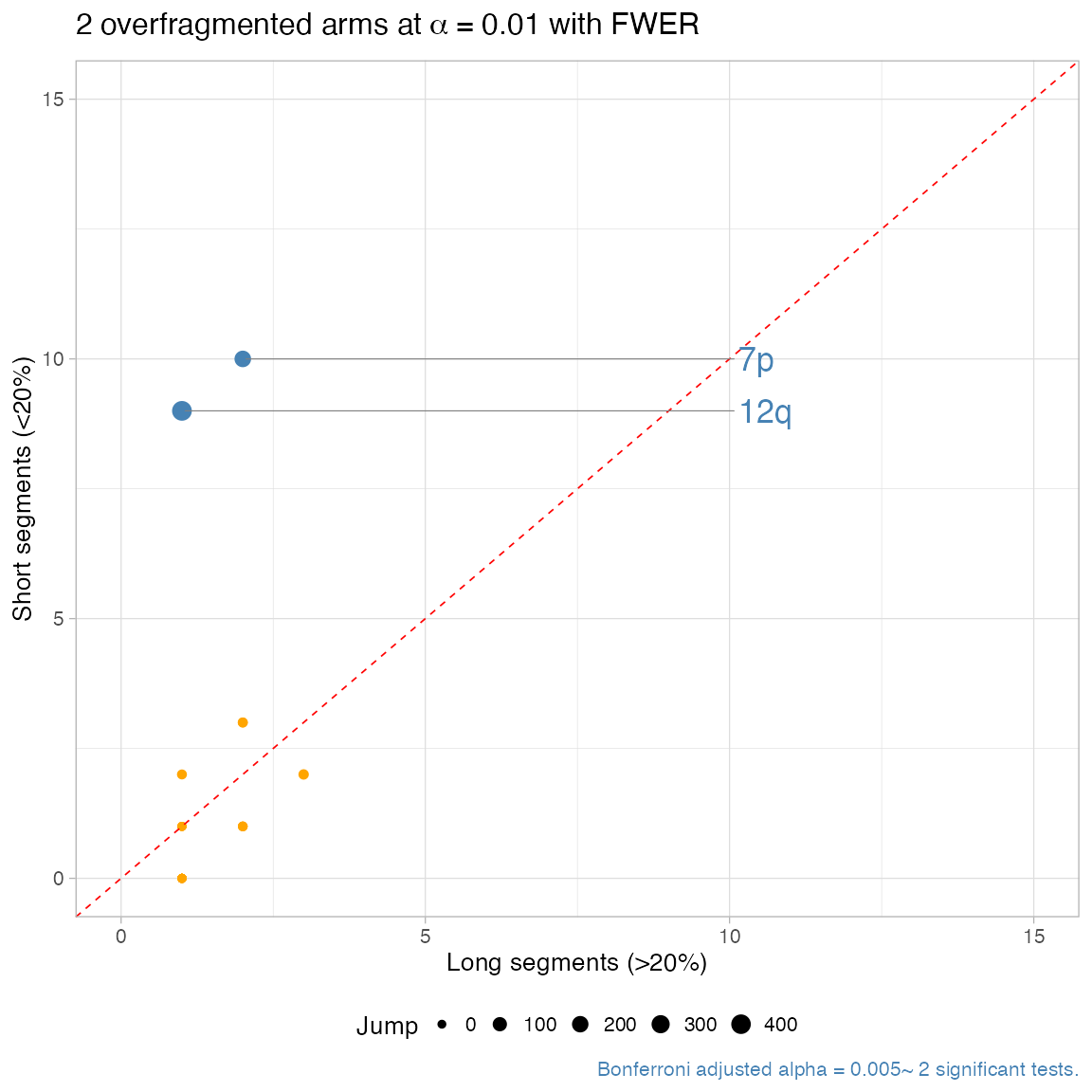
#> ℹ One-tailed Binomial test: 2 tests, alpha 0.01. Short segments: 0.2% of the reference arm.
#> ✔ chr7p, p = 4.52608e-06 ~ 12 segments, 10 short.
#> ✔ chr12q, p = 4.19839999999999e-06 ~ 10 segments, 9 short.
#> ℹ 2 significantly overfragmented chromosome arms (alpha level 0.01).
print(x)
#> ── [ CNAqc ] MySample 12963 mutations in 87 segments (87 clonal, 0 subclonal). G
#>
#> ── Clonal CNAs
#>
#> 2:2 [n = 7478, L = 1493 Mb] ■■■■■■■■■■■■■■■■■■■■■■■■■■■ { CTCF }
#> 4:2 [n = 1893, L = 333 Mb] ■■■■■■■
#> 3:2 [n = 1625, L = 362 Mb] ■■■■■■
#> 2:1 [n = 1563, L = 424 Mb] ■■■■■■ { TTN }
#> 3:0 [n = 312, L = 139 Mb] ■
#> 2:0 [n = 81, L = 39 Mb] { TP53 }
#> 16:2 [n = 4, L = 0 Mb]
#> 25:2 [n = 2, L = 1 Mb]
#> 3:1 [n = 2, L = 1 Mb]
#> 106:1 [n = 1, L = 0 Mb]
#> ℹ Sample Purity: 89% ~ Ploidy: 4.
#> ℹ There are 3 annotated driver(s) mapped to clonal CNAs.
#> chr from to ref alt DP NV VAF driver_label is_driver
#> chr2 179431633 179431634 C T 117 77 0.6581197 TTN TRUE
#> chr16 67646006 67646007 C T 120 54 0.4500000 CTCF TRUE
#> chr17 7577106 7577107 G C 84 78 0.9285714 TP53 TRUE
#> ✔ These segments are smoothed; before smoothing there were 267 segments.
#> ✔ Arm-level fragmentation analysis: 2 segments overfragmented.
plot_arm_fragmentation(x, zoom = 0)