Plot a barplot for the segments, reporting either their counts of the proportion of genome covered.
Arguments
Value
A ggplot2 plot.
Examples
data('example_dataset_CNAqc', package = 'CNAqc')
x = init(mutations = example_dataset_CNAqc$mutations, cna = example_dataset_CNAqc$cna, purity = example_dataset_CNAqc$purity)
#>
#> ── CNAqc - CNA Quality Check ───────────────────────────────────────────────────
#>
#> ℹ Using reference genome coordinates for: GRCh38.
#> ✔ Found annotated driver mutations: TTN, CTCF, and TP53.
#> ✔ Fortified calls for 12963 somatic mutations: 12963 SNVs (100%) and 0 indels.
#> ! CNAs have no CCF, assuming clonal CNAs (CCF = 1).
#> ✔ Fortified CNAs for 267 segments: 267 clonal and 0 subclonal.
#> ✔ 12963 mutations mapped to clonal CNAs.
# All chromosomes (default)
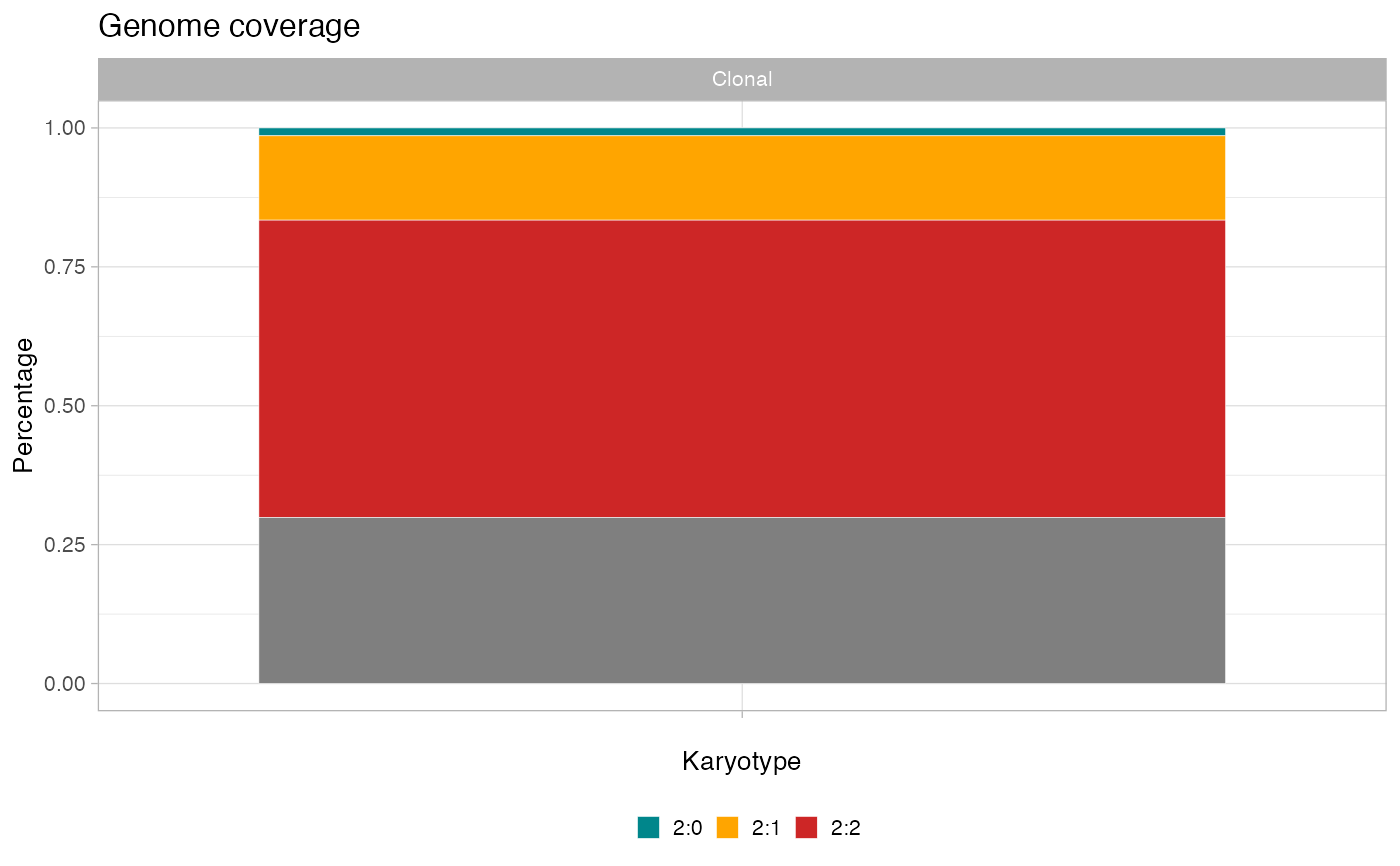
plot_karyotypes(x)
#> Warning: Ignoring unknown parameters: `size`
 # Specific chromosomes
plot_karyotypes(x, chromosomes = 'chr3')
#> Warning: Ignoring unknown parameters: `size`
#> Warning: No shared levels found between `names(values)` of the manual scale and the
#> data's fill values.
#> Warning: No shared levels found between `names(values)` of the manual scale and the
#> data's fill values.
# Specific chromosomes
plot_karyotypes(x, chromosomes = 'chr3')
#> Warning: Ignoring unknown parameters: `size`
#> Warning: No shared levels found between `names(values)` of the manual scale and the
#> data's fill values.
#> Warning: No shared levels found between `names(values)` of the manual scale and the
#> data's fill values.
 plot_karyotypes(x, chromosomes = 'chr13')
#> Warning: Ignoring unknown parameters: `size`
plot_karyotypes(x, chromosomes = 'chr13')
#> Warning: Ignoring unknown parameters: `size`
