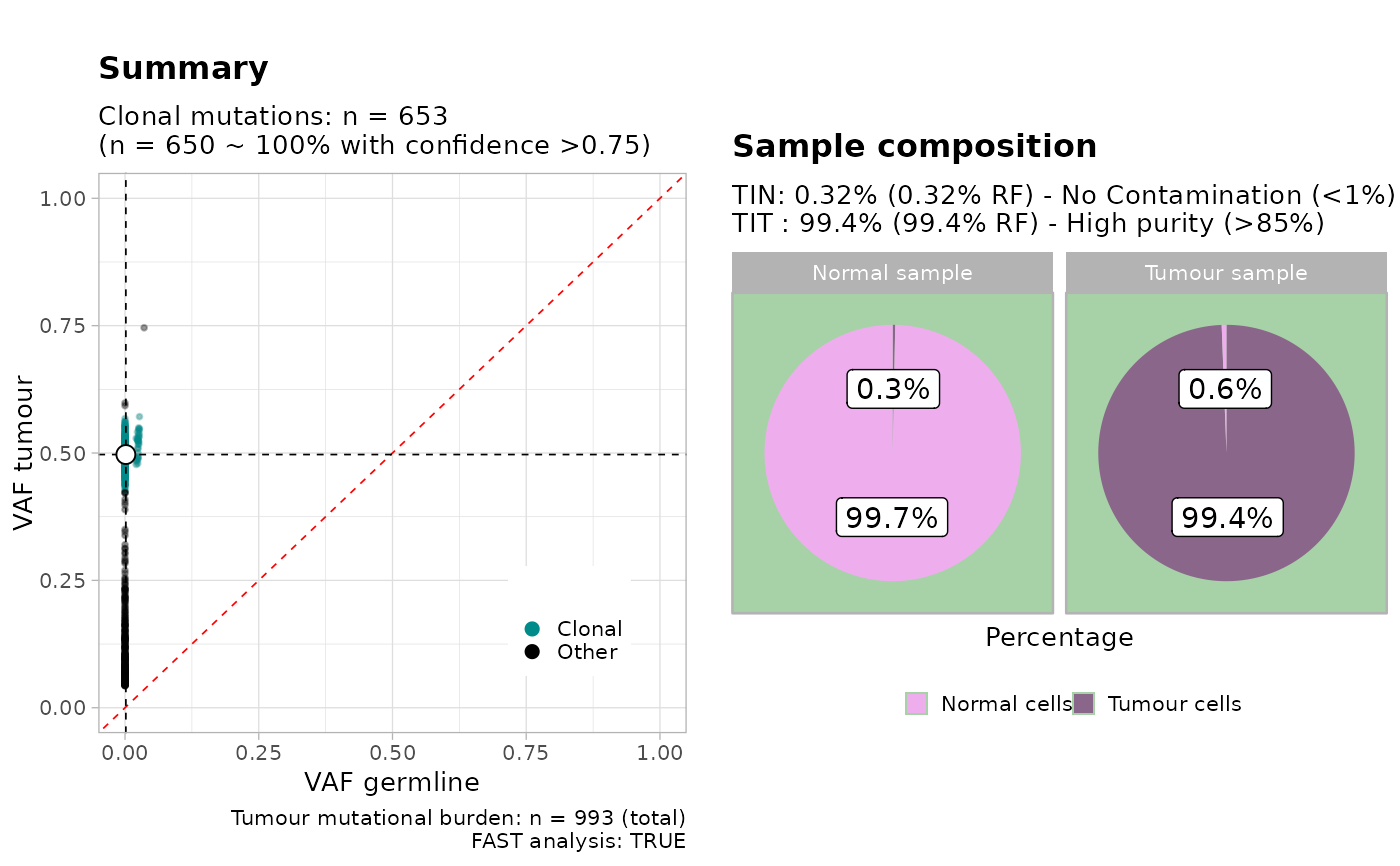
This function is a wrapper to call plot_simple_report.
# S3 method for tin_obj
plot(x, ...)Arguments
- x
A TINC analysis computed with
autofit.- ...
Extra S3 parameters.
Value
A TINC analysis plot (a `ggplot` figure with multiple panels).
See also
plot_simple_report.
Examples
rt = random_TIN()
#> ✔ Generated TINC dataset (n = 994 mutations), TIN (0.05) and TIT (1), normal and tumour coverage 30x and 120x.
#> Warning: Removed 2 rows containing missing values or values outside the scale range
#> (`geom_bar()`).
#> Warning: Removed 2 rows containing missing values or values outside the scale range
#> (`geom_bar()`).
plot(autofit(input = rt$data, cna = rt$cna, FAST = TRUE))
#> [ TINC ]
#>
#>
#> ── Loading TINC input data ─────────────────────────────────────────────────────
#> ✔ Input data contains n = 994 mutations, selecting operation mode.
#> ! Found CNA data, retaining only mutations that map to segments with predominant karyotype ...
#>
#>
#> ── CNAqc - CNA Quality Check ───────────────────────────────────────────────────
#>
#> ℹ Using reference genome coordinates for: GRCh38.
#> ✔ Fortified calls for 994 somatic mutations: 994 SNVs (100%) and 0 indels.
#> ! CNAs have no CCF, assuming clonal CNAs (CCF = 1).
#> ! Added segments length (in basepairs) to CNA segments.
#> ✔ Fortified CNAs for 994 segments: 994 clonal and 0 subclonal.
#> Warning: [CNAqc] a karyotype column is present in CNA calls, and will be overwritten
#> ✔ 994 mutations mapped to clonal CNAs.
#>
#>
#> ── Genome coverage by karyotype, in basepairs. ──
#>
#> # A tibble: 1 × 4
#> minor Major n karyotype
#> <dbl> <dbl> <dbl> <chr>
#> 1 1 1 2982 1:1
#> ✔ n = 994 mutations mapped to CNA segments with karyotype 1:1 (largest available in basepairs).
#> ✔ Mutation with VAF within 0 and 0.7 ~ n = 993.
#>
#> ── Analysing tumour sample with MOBSTER ────────────────────────────────────────
#>
#> [ MOBSTER fit ]
#>
#> ✔ Loaded input data, n = 993.
#> ❯ n = 993. Mixture with k = 1,2 Beta(s). Pareto tail: TRUE and FALSE. Output
#> clusters with π > 0.02 and n > 10.
#> ! mobster automatic setup FAST for the analysis.
#> ❯ Scoring (without parallel) 2 x 2 x 2 = 8 models by reICL.
#>
#>
#>
#> ℹ MOBSTER fits completed in 4.4s.
#>
#> ── [ MOBSTER ] My MOBSTER model n = 993 with k = 1 Beta(s) and a tail ──────────
#> ● Clusters: π = 65% [C1] and 35% [Tail], with π > 0.
#> ● Tail [n = 340, 35%] with alpha = 1.2.
#> ● Beta C1 [n = 653, 65%] with mean = 0.5.
#> ℹ Score(s): NLL = -1367.06; ICL = -2653.92 (-2692.71), H = 38.79 (0). Fit
#> converged by MM in 10 steps.
#>
#> ℹ With CNA, TINC will estimating tumour purity adjusting by copy number and mutation multiplicity.
#> ℹ Mutant allele copies 1 for karyotype 1:1
#> Warning: You did not pass enough input colours, adding a gray colour
#> Available: C1, Tail
#> Missing: NA
#>
#> ✔ MOBSTER found n = 650 clonal mutations from cluster C1
#>
#> ── Analysing normal sample with BMix ───────────────────────────────────────────
#>
#>
#> ── BMix fit ────────────────────────────────────────────────────────────────────
#>
#> ℹ Binomials k_B = 1 and 2, Beta-Binomials k_BB = 0; 4 fits to run.
#>
#> ℹ Bmix best fit completed in 0 mins
#>
#> ── [ BMix ] My BMix model n = 650 with k = 2 component(s) (2 + 0) ──────────────
#> • Clusters: π = 95% [Bin 2] and 5% [Bin 1], with π > 0.
#> • Binomial Bin 1 with mean = 0.00027488445714558.
#> • Binomial Bin 2 with mean = 0.00165701647139773.
#> ℹ Score (model selection): ICL = 384.92.
#> Scale for x is already present.
#> Adding another scale for x, which will replace the existing scale.
#> Scale for fill is already present.
#> Adding another scale for fill, which will replace the existing scale.
#> Warning: Removed 2 rows containing missing values or values outside the scale range
#> (`geom_bar()`).
#> Warning: Removed 551 rows containing missing values or values outside the scale range
#> (`geom_raster()`).
#> ✔ Binomial peaks 0.00027488445714558 and 0.00165701647139773 with proportions 0.0469447884688011 and 0.953055211531199. Clonal score 0.0015921325763527 with TINN 0.00318426515270541
#>
#> ── Analysing tumour and normal samples with VIBER ──────────────────────────────
#>
#> [ VIBER - variational fit ]
#>
#> ℹ Input n = 993, with k < 5. Dirichlet concentration α = 1e-06.
#> ℹ Beta (a_0, b_0) = (1, 1); q_i = prior. Optimise: ε = 1e-06 or 1000 steps, r = 3 starts.
#> [easypar] 2024-04-25 09:23:00.278353 - Overriding parallel execution setup [TRUE] with global option : FALSE
#>
#> ✔ VIBER fit completed in 0.02 mins (status: converged)
#>
#> ── [ VIBER ] My VIBER model n = 993 (w = 2 dimensions). Fit with k = 5 clusters.
#> • Clusters: π = 67% [C2], 28% [C4], and 6% [C1], with π > 0.
#> • Binomials: θ = <0, 0.5> [C2], <0, 0.08> [C4], and <0, 0.22> [C1].
#> ℹ Score(s): ELBO = -136081.912. Fit converged in 49 steps, ε = 1e-06.
#>
#> ✔ Reduced to k = 3 (from 5) selecting VIBER cluster(s) with π > 0.02, and Binomial p > 0 in w > 0 dimension(s).