Analysis pipeline
The TINC pipelines requires to run a single function:
autofit.
Results can then be inspected using two report functions (see below).
Example simulated inputs
TIN provides a function to generate a random dataset to test the
tool. The input TIN and TIT parameters can be
specified.
Note: the sampler generates VAFs using MOBSTER, and then samples some clonal mutations and contaminates the normal. This process is noisy and might return slightly lower values for the actual
TITestimate. The best setup to test TINC is with a simulated BAM file - e.g., created via BAMSurgeon.
set.seed(1234)
# Sample data from a tumour with 80% purity, and 10% contamination in the normal
sampled_data = random_TIN(TIT = .8, TIN = 0.1)
print(sampled_data)
#> $data
#> # A tibble: 984 × 14
#> chr from to ref alt simulated_cluster n_tot_count t_tot_count
#> <chr> <int> <dbl> <chr> <chr> <chr> <int> <int>
#> 1 chr3 141170745 1.41e8 G G C1 25 117
#> 2 chr14 52211493 5.22e7 C G C1 32 108
#> 3 chr19 33758990 3.38e7 C C C1 40 134
#> 4 chr8 139096754 1.39e8 C A C1 23 120
#> 5 chr15 81762079 8.18e7 C G C1 25 136
#> 6 chr14 10406851 1.04e7 C T C1 33 119
#> 7 chr12 59535606 5.95e7 G A C1 26 107
#> 8 chr20 244958 2.45e5 C C C1 27 115
#> 9 chr9 10238563 1.02e7 T A C1 30 117
#> 10 chr17 45642943 4.56e7 C A C1 30 127
#> # ℹ 974 more rows
#> # ℹ 6 more variables: n_alt_count <dbl>, t_alt_count <dbl>, n_ref_count <dbl>,
#> # t_ref_count <dbl>, sim_t_vaf <dbl>, sim_n_vaf <dbl>
#>
#> $cna
#> # A tibble: 984 × 6
#> chr from to ref Major minor
#> <chr> <dbl> <dbl> <chr> <dbl> <dbl>
#> 1 chr3 141170744 141170747 G 1 1
#> 2 chr14 52211492 52211495 C 1 1
#> 3 chr19 33758989 33758992 C 1 1
#> 4 chr8 139096753 139096756 C 1 1
#> 5 chr15 81762078 81762081 C 1 1
#> 6 chr14 10406850 10406853 C 1 1
#> 7 chr12 59535605 59535608 G 1 1
#> 8 chr20 244957 244960 C 1 1
#> 9 chr9 10238562 10238565 T 1 1
#> 10 chr17 45642942 45642945 C 1 1
#> # ℹ 974 more rows
#>
#> $plot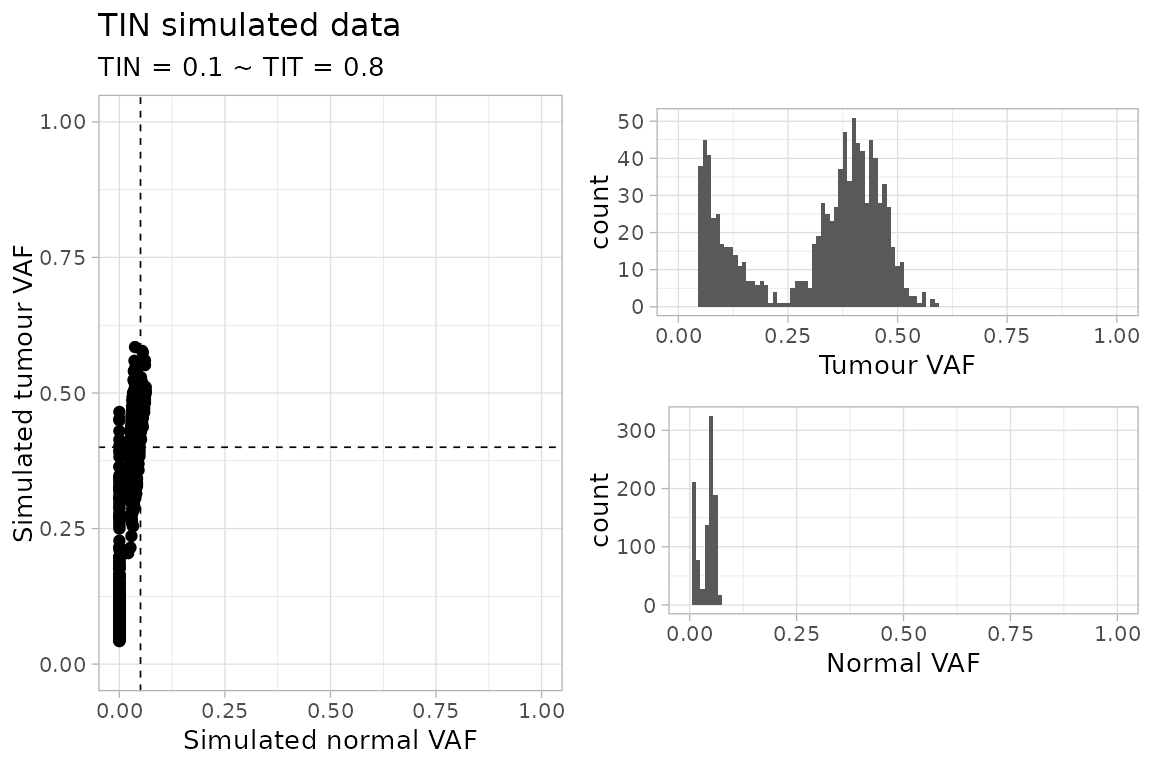
The sampler generates random SNVs, and plain diploid CNA segments. These segments are not real and span just through each one of the simulated SNVs.
Data mappings - done internally by TINC - are also available via the
load_TINC_input function.
load_TINC_input(x = sampled_data$data, cna = sampled_data$cna)
#>
#> ── Loading TINC input data ─────────────────────────────────────────────────────
#> ✔ Input data contains n = 984 mutations, selecting operation mode.
#> ! Found CNA data, retaining only mutations that map to segments with predominant karyotype ...
#>
#> ── CNAqc - CNA Quality Check ───────────────────────────────────────────────────
#> ℹ Using reference genome coordinates for: GRCh38.
#> ✔ Fortified calls for 984 somatic mutations: 984 SNVs (100%) and 0 indels.
#> ! CNAs have no CCF, assuming clonal CNAs (CCF = 1).
#> ! Added segments length (in basepairs) to CNA segments.
#> ✔ Fortified CNAs for 984 segments: 984 clonal and 0 subclonal.
#> Warning in map_mutations_to_clonal_segments(mutations, cna_clonal): [CNAqc] a
#> karyotype column is present in CNA calls, and will be overwritten
#> ✔ 984 mutations mapped to clonal CNAs.
#>
#> ── Genome coverage by karyotype, in basepairs. ──
#>
#> # A tibble: 1 × 4
#> minor Major n karyotype
#> <dbl> <dbl> <dbl> <chr>
#> 1 1 1 2952 1:1
#> ✔ n = 984 mutations mapped to CNA segments with karyotype 1:1 (largest available in basepairs).
#> ✔ Mutation with VAF within 0 and 0.7 ~ n = 984.
#> $mutations
#> # A tibble: 984 × 12
#> chr from to ref alt n_ref_count n_alt_count t_ref_count
#> <chr> <int> <dbl> <chr> <chr> <dbl> <dbl> <dbl>
#> 1 chr3 141170745 141170746 G G 24 1 71
#> 2 chr14 52211493 52211494 C G 31 1 73
#> 3 chr19 33758990 33758991 C C 38 2 74
#> 4 chr8 139096754 139096755 C A 22 1 63
#> 5 chr15 81762079 81762080 C G 24 1 76
#> 6 chr14 10406851 10406852 C T 31 2 60
#> 7 chr12 59535606 59535607 G A 25 1 62
#> 8 chr20 244958 244959 C C 26 1 65
#> 9 chr9 10238563 10238564 T A 29 1 73
#> 10 chr17 45642943 45642944 C A 29 1 79
#> # ℹ 974 more rows
#> # ℹ 4 more variables: t_alt_count <dbl>, karyotype <chr>, id <chr>,
#> # OK_tumour <lgl>
#>
#> $cna_map
#> ── [ CNAqc ] MySample 984 mutations in 984 segments (984 clonal, 0 subclonal). G
#>
#> ── Clonal CNAs
#>
#> 1:1 [n = 984, L = 0 Mb] ■■■■■■■■■■■■■■■■■■■■■■■■■■■
#> ℹ Sample Purity: 80% ~ Ploidy: 2.
#>
#> $what_we_used
#> [1] "1:1"TINC fits the data via the autofit function.
A flag FAST sets faster set of parameters to determine
TIN and TIT scores - we usually this for prototyping analyses and then
turn it to FALSE.
# Autofit function
TINC_fit = autofit(sampled_data$data, cna = NULL, FAST = TRUE)
#> [ TINC ]
#>
#>
#> ── Loading TINC input data ─────────────────────────────────────────────────────
#> ✔ Input data contains n = 984 mutations, selecting operation mode.
#> ✔ Mutation with VAF within 0 and 0.7 ~ n = 984.
#>
#> ── Analysing tumour sample with MOBSTER ────────────────────────────────────────
#>
#> [ MOBSTER fit ]
#>
#> ✔ Loaded input data, n = 984.
#> ❯ n = 984. Mixture with k = 1,2 Beta(s). Pareto tail: TRUE and FALSE. Output
#> clusters with π > 0.02 and n > 10.
#> ! mobster automatic setup FAST for the analysis.
#> ❯ Scoring (without parallel) 2 x 2 x 2 = 8 models by reICL.
#> ℹ MOBSTER fits completed in 5.3s.
#> ── [ MOBSTER ] My MOBSTER model n = 984 with k = 1 Beta(s) and a tail ──────────
#> ● Clusters: π = 67% [C1] and 33% [Tail], with π > 0.
#> ● Tail [n = 302, 33%] with alpha = 1.2.
#> ● Beta C1 [n = 682, 67%] with mean = 0.4.
#> ℹ Score(s): NLL = -906.1; ICL = -1676.77 (-1770.84), H = 94.07 (0). Fit
#> converged by MM in 13 steps.
#> ℹ Using the location likelihood heuristic to inspect mutations' distribution
#> ✔ Cluster C1: 60% of counts in 12 chromosomes (spread over >20% of used chromosomes: 4.4).
#> ℹ Without CNA, TINC will estimate tumour purity as 2*x, with x the clonal peak.
#> ✔ MOBSTER found n = 664 clonal mutations from cluster C1
#>
#> ── Analysing normal sample with BMix ───────────────────────────────────────────
#>
#> ── BMix fit ────────────────────────────────────────────────────────────────────
#> ℹ Binomials k_B = 1 and 2, Beta-Binomials k_BB = 0; 4 fits to run.
#> ℹ Bmix best fit completed in 0 mins
#> ── [ BMix ] My BMix model n = 664 with k = 1 component(s) (1 + 0) ──────────────
#> • Clusters: π = 100% [Bin 1], with π > 0.
#> • Binomial Bin 1 with mean = 0.035546715626275.
#> ℹ Score (model selection): ICL = 1474.26.
#> Scale for x is already present.
#> Adding another scale for x, which will replace the existing scale.
#> Scale for fill is already present.
#> Adding another scale for fill, which will replace the existing scale.
#> ✔ Binomial peaks 0.035546715626275 with proportions 1. Clonal score 0.035546715626275 with TINN 0.07109343125255
#>
#>
#>
#> ── Analysing tumour and normal samples with VIBER ──────────────────────────────
#>
#> [ VIBER - variational fit ]
#>
#> ℹ Input n = 984, with k < 5. Dirichlet concentration α = 1e-06.
#> ℹ Beta (a_0, b_0) = (1, 1); q_i = prior. Optimise: ε = 1e-06 or 1000 steps, r = 3 starts.
#> [easypar] 2024-04-25 09:23:26.800961 - Overriding parallel execution setup [TRUE] with global option : FALSE
#> ✔ VIBER fit completed in 0.01 mins (status: converged)
#> ── [ VIBER ] My VIBER model n = 984 (w = 2 dimensions). Fit with k = 5 clusters.
#> • Clusters: π = 70% [C4] and 30% [C2], with π > 0.
#> • Binomials: θ = <0.03, 0.4> [C4] and <0, 0.09> [C2].
#> ℹ Score(s): ELBO = -137271.765. Fit converged in 24 steps, ε = 1e-06.
#> ✔ Reduced to k = 2 (from 5) selecting VIBER cluster(s) with π > 0.02, and Binomial p > 0 in w > 0 dimension(s).Summary of the results (with an S3 object)
print(TINC_fit)
#>
#> ── TINC profiler for bulk WGS ──────────────────────────────────────────────────
#> ! Copy Number data has not been used for this analysis.
#> → Mutations data: n = 984 out of 984 within range (100%).
#>
#> TIT : 80% (RF 80) ~ n = 664 clonal mutations, cluster C1
#> TIN : 7% (RF 7) ~ n = 633 with VAF > 0
#>
#> QC Tumour Good purity (65-85%)
#> QC Normal Contamination (7-15%)Plot the results (with an S3 object).
plot(TINC_fit)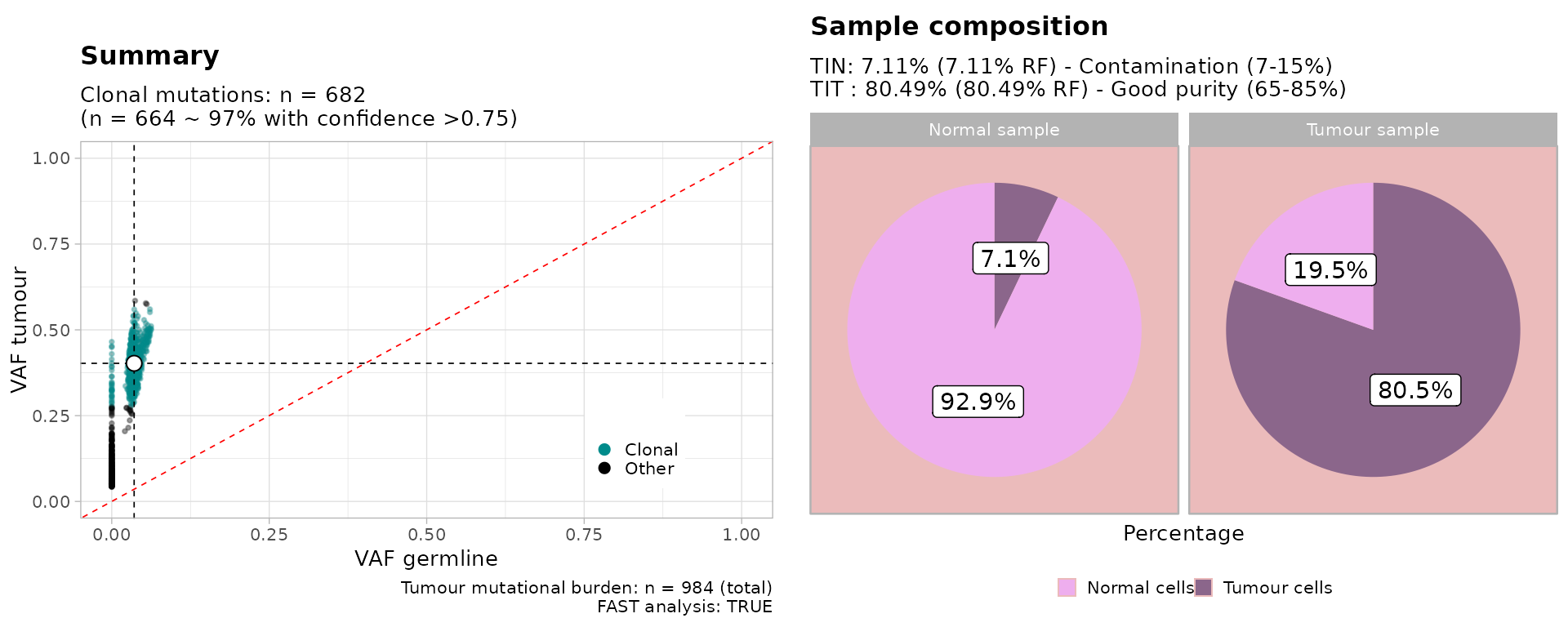
# Analogous to
# plot_simple_report(TINC_fit)Or make a more detailed report
plot_full_page_report(TINC_fit)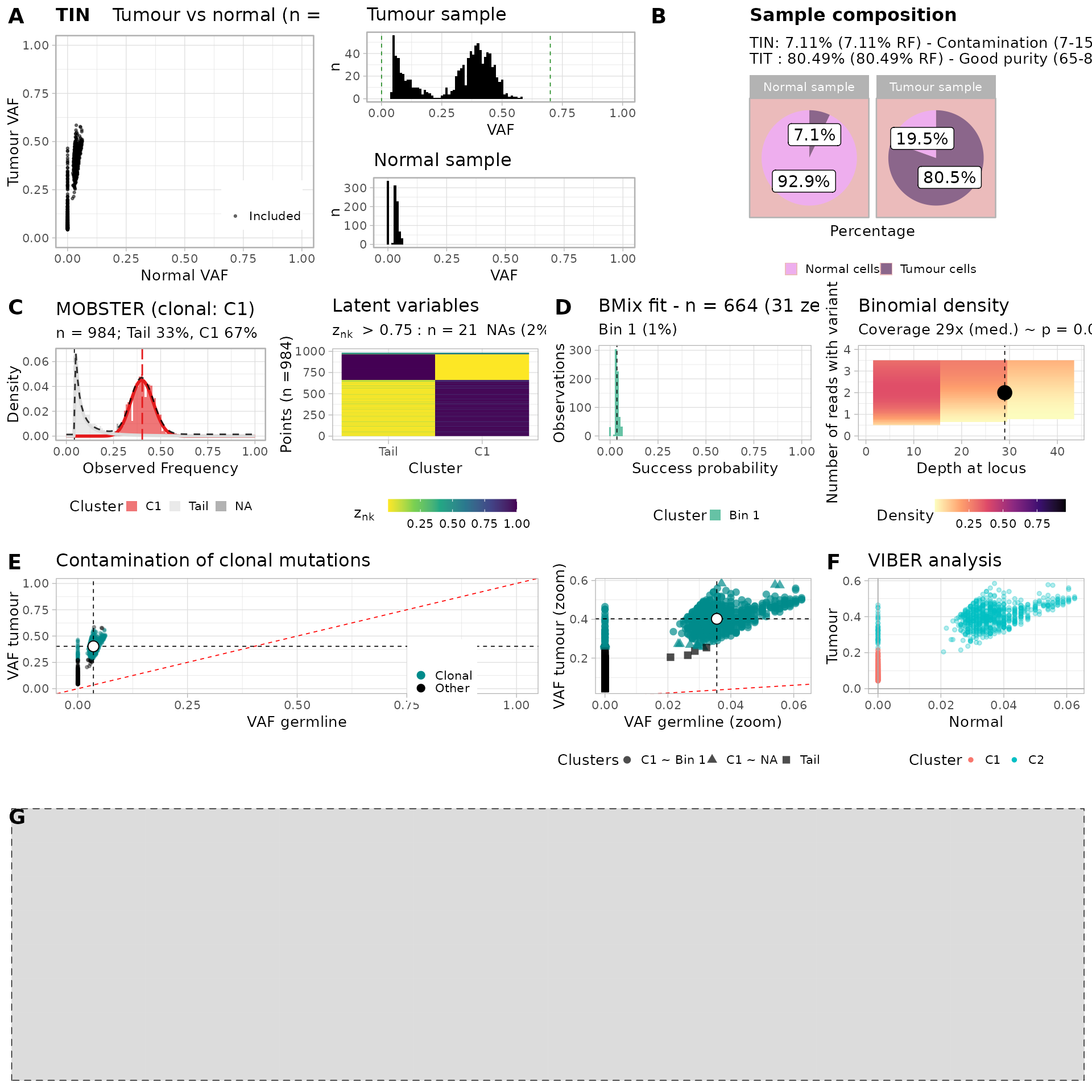
A similar fit object is available inside the package (called
fit_example).