ProCESS wraps the CLONES simulation and data-generation engine by using the R/C++ Rcpp interface, making it easy to “program” stochastic tumour evolution.
Pipeline overview

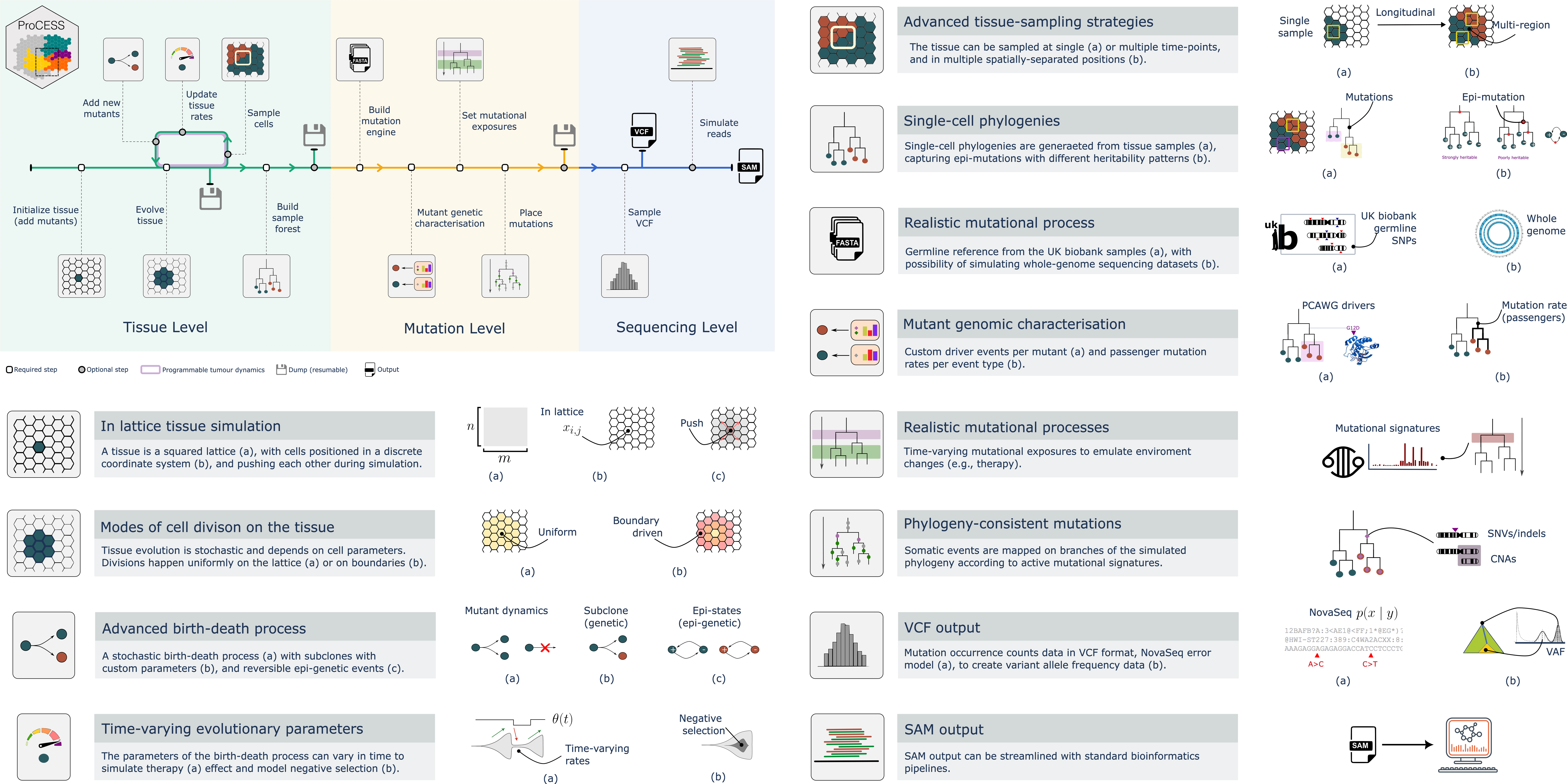
ProCESS’s pipeline consists of three steps.
a discrete tissue is first simulated, with distinct clones defined by stochastic rates of growth and death. These compete to colonise the tissue in a standard stochastic birth-death process, which in ProCESS is empowered by a time-varying structure that enables easily modelling of complex evolutionary forces that vary over time (e.g., therapy-induced negative selection). The tissue can be sampled at multiple time-points and in multiple spatially separated positions.
from the sampled cells, the simulated phylogenetic history of the tumour can be extracted. The distance among the involved cells will represent the lineage history of the simulated clones, and mutational processes can be used to attach genomes to each simulated cell. ProCESS supports standard germline and somatic mutations, including single-nucleotide variants (SNVs), insertion-deletions (indels) and copy number alterations (CNAs). For SNVs and indels, time-varying mutational signatures can also be included.
from the simulated phylogenetic history of the tumour, it is possible to generate tumour-matched-normal synthetic data for benchmarking bioinformatics tools, or to infer parameters against real sequencing data. ProCESS output includes common VCF formats, as well as low-level SAM outputs for alignment and downstream analysis. Every simulated sample can be assigned custom coverage and purity.
Main features
The ProCESS engine has a number of features, the most relevant of which are:
- Cells are associated with species that proliferate in a stochastic fashion on a 2D lattice.
- Every species is defined by (i) an abstract “mutant” and (ii) an epi-state; the epi-state is binary (0/1, on-off). The combination of the mutant and the epi-state determines the evolutionary parameters of each species, which can change over time mimicking treatment-related evolutionary pressures, for instance.
- Species evolution is stochastic and follows a no-back mutation model, where at every cell division the mutant status is heritable, while the epi-state can be stochastically reversible.
- At the molecular level, the genomes of tumour cells are mutated by simple mutations (SNVs and indels) and more complex CNAs. Mutations - in the broad sense - can be either passenger or driver ones, and can be linked to realistic mutational processes. All their rates of accumulation can change over time, mimicking the emergence of time-varying mutational forces (e.g., to model treatment). Moreover, realistic germline samples can be simulated by interfacing with the UK bio-bank database.
- Arbitrary tissue sampling schema can be simulated, including multi-region and longitudinal datasets with any number of samples and time-points.
- Realistic read-counts based on bulk sequencing data can be generated, both at the level of whole-genome, whole-exome and targeted panels, as well as at the level of pre-processed outputs (VCFs) or low-level sequencing outputs (SAM).
- Full access to the evolutionary process and its output is available, allowing easy testing of complex clonal architecture identification methods, mutation callers, etc.
Detailed pipeline
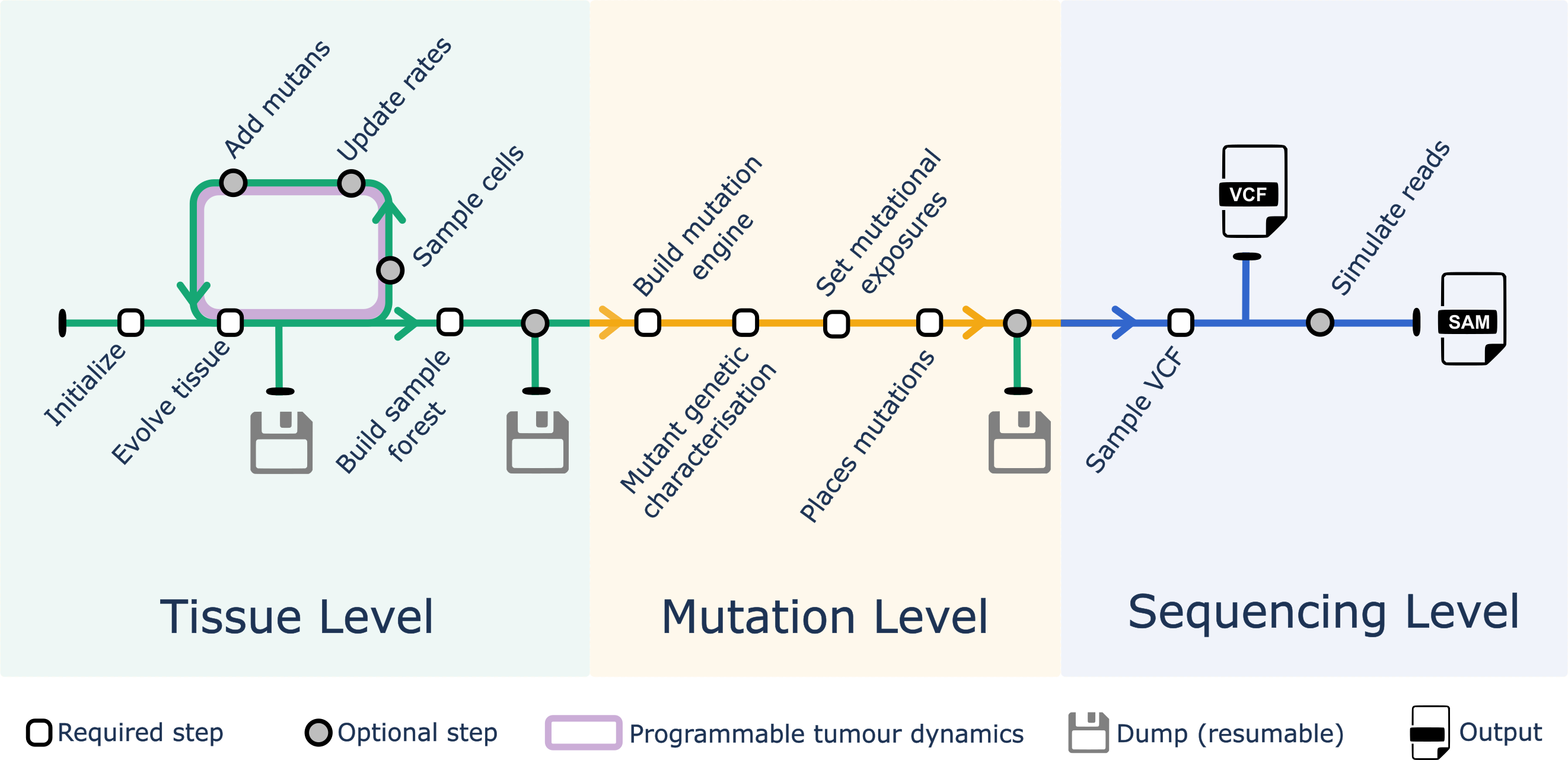
A ProCESS simulation follows the steps (some required, some optional) that we discussed below. The state of the simulation can be saved (and resumed) at several time-points.
Tissue simulation
- Initialisation: a rectangular tissue is initialised, some species are defined, and a few cells are placed onto it.
- Tissue evolution: all the cells in the tissue are left to grow stochastically.
- Sample cells: cells can be sampled from the tissue to mimic a measurement.
- Updates/add species: the parameters of each species can be modified, and cells from new species can be defined and added to the tissue (subclones).
Steps 2-4 represent an iterative interface for “programmable tumour dynamics” that gives the user the flexibility to code a custom evolutionary process.
Mutations generation
- Phylogeny: from sampled cells, a phylogeny is built that reflects the evolutionary history of the simulated tumour.
- Mutational processes: mutational processes can be mapped onto the temporal evolution of the process.
- Mutational engine: an engine for simulating mutations is built from a real reference genome.
- Map mutations: mutations are stochastically attached to the simulated phylogeny.
Sequencing data generation
- VCF: given mutations on the phylogenetic tree, custom coverage and sample purity, a VCF can be simulated to mimic variant-calling with Beta-Binomial noise.
- SAM: from the same mutations. it is possible to generate reads for a tumour-matched-normal assay, and feed those data into a custom bioinformatics pipeline.