2. Inference of mutation copy number and multiplicity
Source:vignettes/a2_classify_mutations.Rmd
a2_classify_mutations.Rmd
library(INCOMMON)
#> Warning: replacing previous import 'cli::num_ansi_colors' by
#> 'crayon::num_ansi_colors' when loading 'INCOMMON'
library(dplyr)
#>
#> Attaching package: 'dplyr'
#> The following objects are masked from 'package:stats':
#>
#> filter, lag
#> The following objects are masked from 'package:base':
#>
#> intersect, setdiff, setequal, union
library(DT)
library(cli)
library(crayon)
#>
#> Attaching package: 'crayon'
#> The following object is masked from 'package:cli':
#>
#> num_ansi_colorsIn this vignette, we use INCOMMON to infer the copy number and multiplicity of mutations from a single sample of the MSK-MetTropsim dataset provided within the package.
2.1 Input preparation
The minimal input for INCOMMON analyses consists of two pieces.
2.1.1 Genomic data
First we need a table of genomic_data (mutations) with
required columns chr, from, to,
ref, alt, DP, NV,
VAF, and gene.
The following example is taken from the internal dataset obtained from the MSK-MetTropism cohort:
2.1.2 Clinical data
Second, we need a table of clinical data with at least the columns
sample (sample names matching the ones in
genomic_data), purity (purity of each sample)
and tumor_type (annotated tumour type of the sample),
required for using tumour-specific priors. The following example is
taken from the internal dataset obtained from the MSK-MetTropism
cohort:
2.1.3 Initialisation of the input
The first thing to do is to initialise the input so that we will have
it in INCOMMON format, through function init. This function
takes as input the tables of genomic_data and
clinical_data, plus optionally, a list of gene roles.
INCOMMON provides a default list cancer_gene_census
obtained from the COSMIC Cancer Gene Census v.98. The required format is
as following:
We now can create the input INCOMMON object through the function
init:
x = init(genomic_data = MSK_genomic_data,
clinical_data = MSK_clinical_data,
gene_roles = cancer_gene_census)
#> ── INCOMMON - Inference of copy number and mutation multiplicity in oncology ───
#>
#> ── Genomic data ──
#>
#> ✔ Found 25659 samples, with 224939 mutations in 491 genes
#> ! No read counts found for 1393 mutations in 1393 samples
#> ! Gene name not provided for 1393 mutations
#> ! 201 genes could not be assigned a role (TSG or oncogene)
#>
#> ── Clinical data ──
#>
#> ℹ Provided clinical features:
#> ✔ sample (required for classification)
#> ✔ purity (required for classification)
#> ✔ tumor_type
#> ✔ OS_MONTHS
#> ✔ OS_STATUS
#> ✔ SAMPLE_TYPE
#> ✔ MET_COUNT
#> ✔ METASTATIC_SITE
#> ✔ MET_SITE_COUNT
#> ✔ PRIMARY_SITE
#> ✔ SUBTYPE_ABBREVIATION
#> ✔ GENE_PANEL
#> ✔ SEX
#> ✔ TMB_NONSYNONYMOUS
#> ✔ FGA
#> ✔ AGE_AT_SEQUENCING
#> ✔ RACE
#> ✔ Found 25257 matching samples
#> ✖ Found 513 unmatched samples
print(x)
#> ── [ INCOMMON ] 223546 PASS mutations across 24266 samples,
#> with 490 mutant gen
#> ℹ Average sample purity: 0.4
#> ℹ Average sequencing depth: 660
#> # A tibble: 223,546 × 27
#> sample tumor_type purity chr from to ref alt DP NV VAF
#> <chr> <chr> <dbl> <chr> <dbl> <dbl> <chr> <chr> <int> <int> <dbl>
#> 1 P-0028912 CHOL 0.3 chr17 7.58e6 7.58e6 G A 837 133 0.159
#> 2 P-0028912 CHOL 0.3 chr6 1.12e8 1.12e8 - A 698 141 0.202
#> 3 P-0028912 CHOL 0.3 chrX 5.32e7 5.32e7 G A 832 85 0.102
#> 4 P-0003698 BLCA 0.2 chr17 7.58e6 7.58e6 C A 437 109 0.249
#> 5 P-0003698 BLCA 0.2 chr3 4.99e7 4.99e7 C A 591 86 0.146
#> 6 P-0003698 BLCA 0.2 chr5 1.49e8 1.49e8 C T 360 36 0.1
#> 7 P-0003698 BLCA 0.2 chr13 3.29e7 3.29e7 G C 1027 162 0.158
#> 8 P-0003698 BLCA 0.2 chr13 3.29e7 3.29e7 G C 1021 182 0.178
#> 9 P-0003698 BLCA 0.2 chr19 1.11e7 1.11e7 G T 573 98 0.171
#> 10 P-0003698 BLCA 0.2 chr22 4.15e7 4.15e7 G A 416 45 0.108
#> # ℹ 223,536 more rows
#> # ℹ 16 more variables: gene <chr>, gene_role <chr>, OS_MONTHS <dbl>,
#> # OS_STATUS <dbl>, SAMPLE_TYPE <chr>, MET_COUNT <dbl>, METASTATIC_SITE <chr>,
#> # MET_SITE_COUNT <dbl>, PRIMARY_SITE <chr>, SUBTYPE_ABBREVIATION <chr>,
#> # GENE_PANEL <chr>, SEX <chr>, TMB_NONSYNONYMOUS <dbl>, FGA <dbl>,
#> # AGE_AT_SEQUENCING <dbl>, RACE <chr>The MSK-MET dataset comprises 25257 samples with matched clinical data. The average sequencing depth is 649 and the average sample purity is 0.4. The 175054 mutations flagged as PASS are the ones that satisfy the requirements for INCOMMON classification: available and non-negative sample purity; integer sequencing depth and number of reads with the variant, character gene names etc.
2.2 Inference of copy number and mutation multiplicity
In principle, INCOMMON can infer any configuration of total copy
number k and mutation multiplicity m, given
that both k and m are integer numbers and
.
Since there is an infinite number of such possible configurations, a
maximum value k_max of k, expected to be found
in the dataset, must be set up prior to the classification task. By
default, INCOMMON uses
.
2.2.1 Rate of read counts per chromosome copy
INCOMMON is a Bayesian model that infers mutation copy number and
multiplicity from read counts. An essential parameter of the model is
the rate of read counts per chromosome copy
.
To guide the inference of this parameter, we use a Gamma prior
distribution. The only a priori information that we have access to is on
the
configurations of mutant genes. We use this to compute a prior
distribution over
from the dataset itself, using the
functioncompute_eta_prior
data('priors_pcawg_hmf')
priors_eta = compute_eta_prior(x = x, priors_k_m = priors_pcawg_hmf)
print(priors_eta)
#> # A tibble: 12 × 6
#> tumor_type mean_eta var_eta N alpha_eta beta_eta
#> <chr> <dbl> <dbl> <int> <dbl> <dbl>
#> 1 BRCA 319. 30947. 2283 3.28 0.0103
#> 2 ESCA 355. 29910. 289 4.21 0.0119
#> 3 GIST 340. 61998. 299 1.87 0.00549
#> 4 HCC 303. 16680. 175 5.50 0.0182
#> 5 LUAD 351. 33989. 3412 3.63 0.0103
#> 6 MEL 306. 29548. 1044 3.17 0.0104
#> 7 OV 314. 19617. 937 5.04 0.0160
#> 8 PAAD 333. 15043. 1698 7.37 0.0221
#> 9 PRAD 283. 20687. 283 3.88 0.0137
#> 10 SCLC 372. 45882. 255 3.01 0.00811
#> 11 STAD 291. 16017. 97 5.27 0.0181
#> 12 PANCA 331. 29117. 10838 3.75 0.0114For each tumour type in the dataset, we estimated the empirical mean and variance of , which can be straightforwardly transformed into the shape parameters and of a Gamma distribution.
We can visualise the prior distribution for each tumour type using
the function plot_eta_prior:
plot_eta_prior(x = x, priors_eta = priors_eta) The plot shows, for each tumour type and at the pan-cancer level
(PANCA), the distribution of total read counts, potentially confused by
diverse copy-number configurations and sample purities, and the
underlying prior distribution of the rate of read counts per chromosome
copy.
The plot shows, for each tumour type and at the pan-cancer level
(PANCA), the distribution of total read counts, potentially confused by
diverse copy-number configurations and sample purities, and the
underlying prior distribution of the rate of read counts per chromosome
copy.
2.2.2 Inference in sample ‘P-0002081’
We now focus on a specific sample:
sample = 'P-0002081'
x = subset_sample(x = x, sample_list = c(sample))
print(x)
#> ── [ INCOMMON ] 4 PASS mutations across 1 samples,
#> with 4 mutant genes across 7
#> ℹ Average sample purity: 0.4
#> ℹ Average sequencing depth: 660
#> # A tibble: 4 × 27
#> sample tumor_type purity chr from to ref alt DP NV VAF
#> <chr> <chr> <dbl> <chr> <dbl> <dbl> <chr> <chr> <int> <int> <dbl>
#> 1 P-0002081 LUAD 0.6 chr12 2.54e7 2.54e7 C A 743 378 0.509
#> 2 P-0002081 LUAD 0.6 chr17 7.58e6 7.58e6 G A 246 116 0.472
#> 3 P-0002081 LUAD 0.6 chr19 1.22e6 1.22e6 C A 260 122 0.469
#> 4 P-0002081 LUAD 0.6 chr19 1.11e7 1.11e7 - C 271 133 0.491
#> # ℹ 16 more variables: gene <chr>, gene_role <chr>, OS_MONTHS <dbl>,
#> # OS_STATUS <dbl>, SAMPLE_TYPE <chr>, MET_COUNT <dbl>, METASTATIC_SITE <chr>,
#> # MET_SITE_COUNT <dbl>, PRIMARY_SITE <chr>, SUBTYPE_ABBREVIATION <chr>,
#> # GENE_PANEL <chr>, SEX <chr>, TMB_NONSYNONYMOUS <dbl>, FGA <dbl>,
#> # AGE_AT_SEQUENCING <dbl>, RACE <chr>The input data table contains 4 mutations affecting KRAS, TP53, STK11 and SMARCA4 genes. In this sample, all 4 mutations have all the required information. We can see that this is a sample of a metastatic lung adenocarcinoma (LUAD), sequenced through the MSK-IMPACT targeted panel version 341, with an estimated purity of 0.6.
INCOMMON models the sample purity probabilistically, centering a Beta
prior around the estimate provided with each sample (usually from a
histopathological assay). The variance
of the distribution must be fixed prior to the classification task
through the argument purity_error. By default, INCOMMON
uses
,
accounting for uncertainty values of around
,
depending on the mean.
The prior purity distribution for a sample can be visualised using
the function plot_purity_prior.
plot_purity_prior(x = x, sample = sample, purity_error = 0.05)
#> Warning: Using `size` aesthetic for lines was deprecated in ggplot2 3.4.0.
#> ℹ Please use `linewidth` instead.
#> ℹ The deprecated feature was likely used in the INCOMMON package.
#> Please report the issue at <https://github.com/caravagnalab/INCOMMON/issues>.
#> This warning is displayed once per session.
#> Call `lifecycle::last_lifecycle_warnings()` to see where this warning was
#> generated.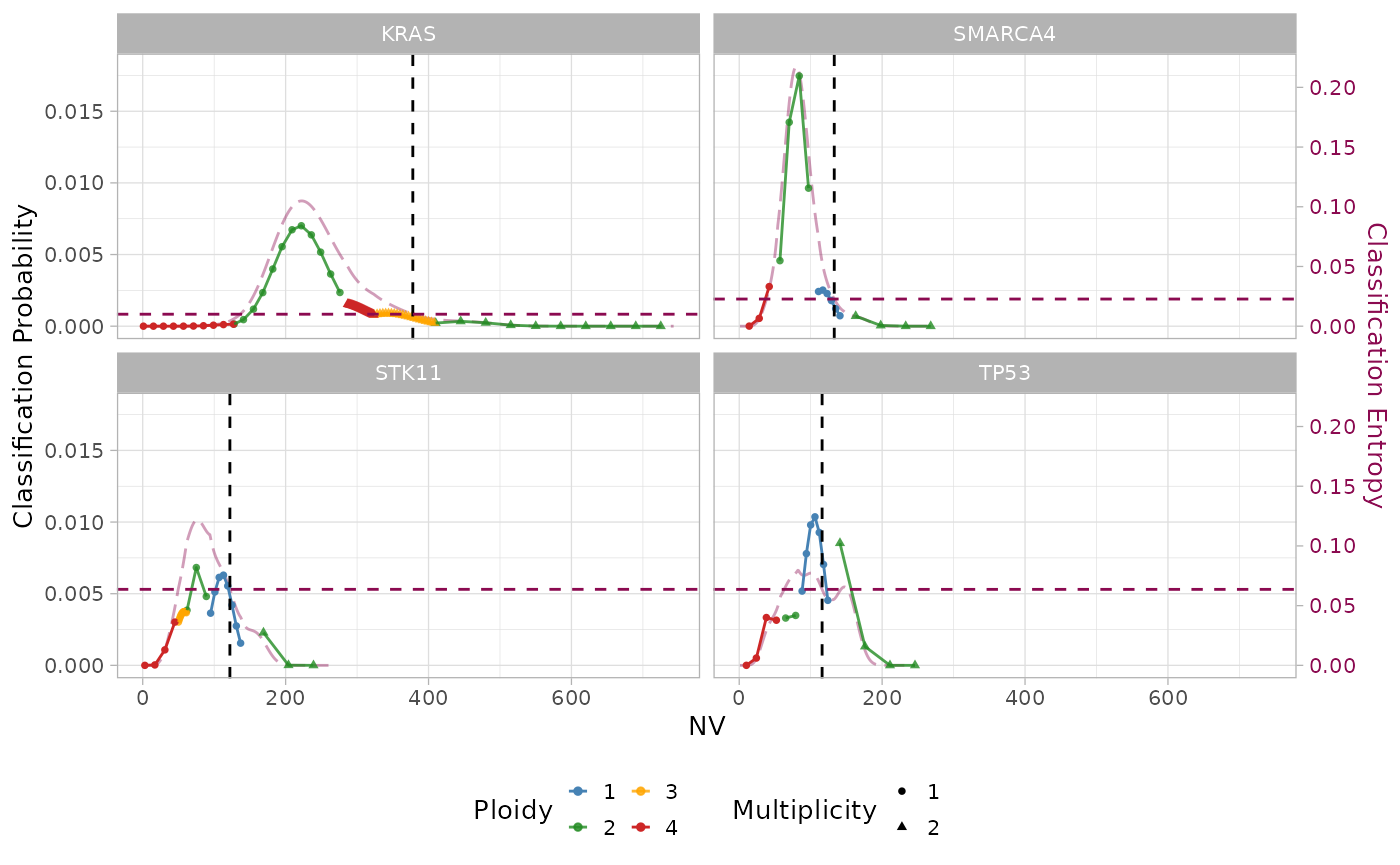
The shape parameters of the Beta distribution ensure that the mean and the variance correspond to the values we provided. The dashed line indicate the input purity estimate, used as the mean of the distribution.
We are now ready to run the classification step through function
classify, using the priors we obtained for
configurations, read count rate per chromosome copy
and sample purity
.
We must provide the number of CPU cores num_cores we want
to use for the parallel MCMC sampling chains, as well as the number of
iterations for the warm-up (iter_warmup) and the proper
sampling (iter_sampling) steps. We also must specify paths
for the directories where we want to store the results
(results_dir), the stan fit objects
(stan_fit_dir) in case we want to store them
(stan_fit_dump = TRUE) and, in case we want to generate a
reporting summary plot (generate_report_plot = TRUE), the
directory where to store the images (reports_dir).
classifyfits the model withcmdstanr, which is not installed automatically with INCOMMON. If you haven’t set it up yet, runinstall.packages("cmdstanr", repos = c("https://mc-stan.org/r-packages/", getOption("repos")))followed bycmdstanr::install_cmdstan()before continuing, or use the INCOMMON web app instead, which requires no local installation.
out = classify(
x = x,
k_max = 8,
priors_k_m = priors_pcawg_hmf,
priors_eta = priors_eta,
purity_error = 0.05,
num_cores = 4,
iter_warmup = 1000,
iter_sampling = 2000,
num_chains = 4
)
print(out)
#> ── [ INCOMMON ] 4 PASS mutations across 1 samples,
#> with 4 mutant genes across 1
#> ℹ Average sample purity: 0.6
#> ℹ Average sequencing depth: 380
#> # A tibble: 4 × 36
#> sample tumor_type purity purity_map eta_map chr from to gene ref
#> <chr> <chr> <dbl> <dbl> <dbl> <chr> <dbl> <dbl> <chr> <chr>
#> 1 P-0002081 LUAD 0.6 0.645 190. chr12 2.54e7 2.54e7 KRAS C
#> 2 P-0002081 LUAD 0.6 0.645 190. chr17 7.58e6 7.58e6 TP53 G
#> 3 P-0002081 LUAD 0.6 0.645 190. chr19 1.22e6 1.22e6 STK11 C
#> 4 P-0002081 LUAD 0.6 0.645 190. chr19 1.11e7 1.11e7 SMAR… -
#> # ℹ 26 more variables: alt <chr>, NV <int>, DP <int>, VAF <dbl>, map_k <int>,
#> # map_m <int>, gene_role <chr>, OS_MONTHS <dbl>, OS_STATUS <dbl>,
#> # SAMPLE_TYPE <chr>, MET_COUNT <dbl>, METASTATIC_SITE <chr>,
#> # MET_SITE_COUNT <dbl>, PRIMARY_SITE <chr>, SUBTYPE_ABBREVIATION <chr>,
#> # GENE_PANEL <chr>, SEX <chr>, TMB_NONSYNONYMOUS <dbl>, FGA <dbl>,
#> # AGE_AT_SEQUENCING <dbl>, RACE <chr>, bayes_p_purity <dbl>,
#> # bayes_p_eta <dbl>, post_pred_p.value_DP <dbl>, …The output contains the maximum-a-posteriori (MAP) values of sample
purity (purity_map), which is close to the input value
(0.578 vs 0.6), read count rate per chromosome copy
(eta_map) around 163, total copy number
(map_k) and multiplicity (map_m).
The only mutant oncogene in the sample is KRAS, that is found in 8 copies, 5 of which mutated, whereas all the TSGs (TP53, SMARCA4, STK11) are in LOH, with only 1 mutant copy and total loss of the WT allele. These inferred values are consistent with the read counts with and without the variant and the estimated proportions of normal and tumour cells in the sample.
2.3 Visualising INCOMMON inference
Since INCOMMON is a Bayesian method, it provides more than point estimates for the quantities it infers. Instead, a full posterior distribution is evaluated.
For example, here we visualise the posterior distribution of copy
number and multiplicity for the 4 mutant genes found in the analysed
sample, using the function plot_posterior_k_m.
plot_posterior_k_m(x = out, k_max = out$parameters$k_max)
For KRAS, most of the probability mass is located at configurations of high total copy number with gain of the mutant allele. Conversely for the TSGs SMARCA4, STK11 and TP53, the posterior probability tends to accumulate on configurations of low total copy number with loss of the WT allele. The red markers indicate the MAP values reported in the above section.