1. Prior distribution of INCOMMON classes from PCAWG
Source:vignettes/a1_pcawg_priors.Rmd
a1_pcawg_priors.Rmd
library(INCOMMON)
#> Warning: replacing previous import 'cli::num_ansi_colors' by
#> 'crayon::num_ansi_colors' when loading 'INCOMMON'
library(dplyr)
library(DT)The inference of copy number and multiplicity of a mutation from read counts only can be much of a hard task, especially in cases where the sample purity or the sequencing depth at the mutation site are low.
For this reason, INCOMMON allows using a prior distribution to improve classifications.
1.1 Empirical priors from PCAWG
When classifying mutations on a specific gene and in samples of a specific tumour type, a categorical prior distribution , where is the ploidy and the mutation multiplicity, can be used to obtain more confident classifications, given that the prior probability of each class is obtained from reliable copy number calls. By default, INCOMMON relies on prior probability obtained from PCAWG whole genomes. From a set of high-confidence copy number calls validated by quality control, we obtained for each gene as the frequency of the corresponding INCOMMON class.
data("pcawg_priors")The empirical priors from PCAWG are provided as an internal data
table pcawg_priors and have the following format
where label represents the lower-level INCOMMON class in
the format <p> N (Mutated: <m> N) and
p is the corresponding value of
.
1.2 User-defined priors
The user who may want to leverage priors obtained in a different way
(e.g. from other datasets or for a specific gene or tumour type not
included in pcawg_priors), can easily do that by creating a
similar data table.
For example:
my_priors = tibble(gene = 'my_gene',
tumor_type = 'my_tumor_type',
label = c("1N (Mutated: 1N)",
"2N (Mutated: 1N)",
"2N (Mutated: 2N)",
"3N (Mutated: 1N)",
"3N (Mutated: 2N)",
"4N (Mutated: 1N)",
"4N (Mutated: 2N)"),
p = c(0.2,0.3,0.1,0.1,0.1,0.1,0.1))The only requirement is that the probabilities sum up to one .
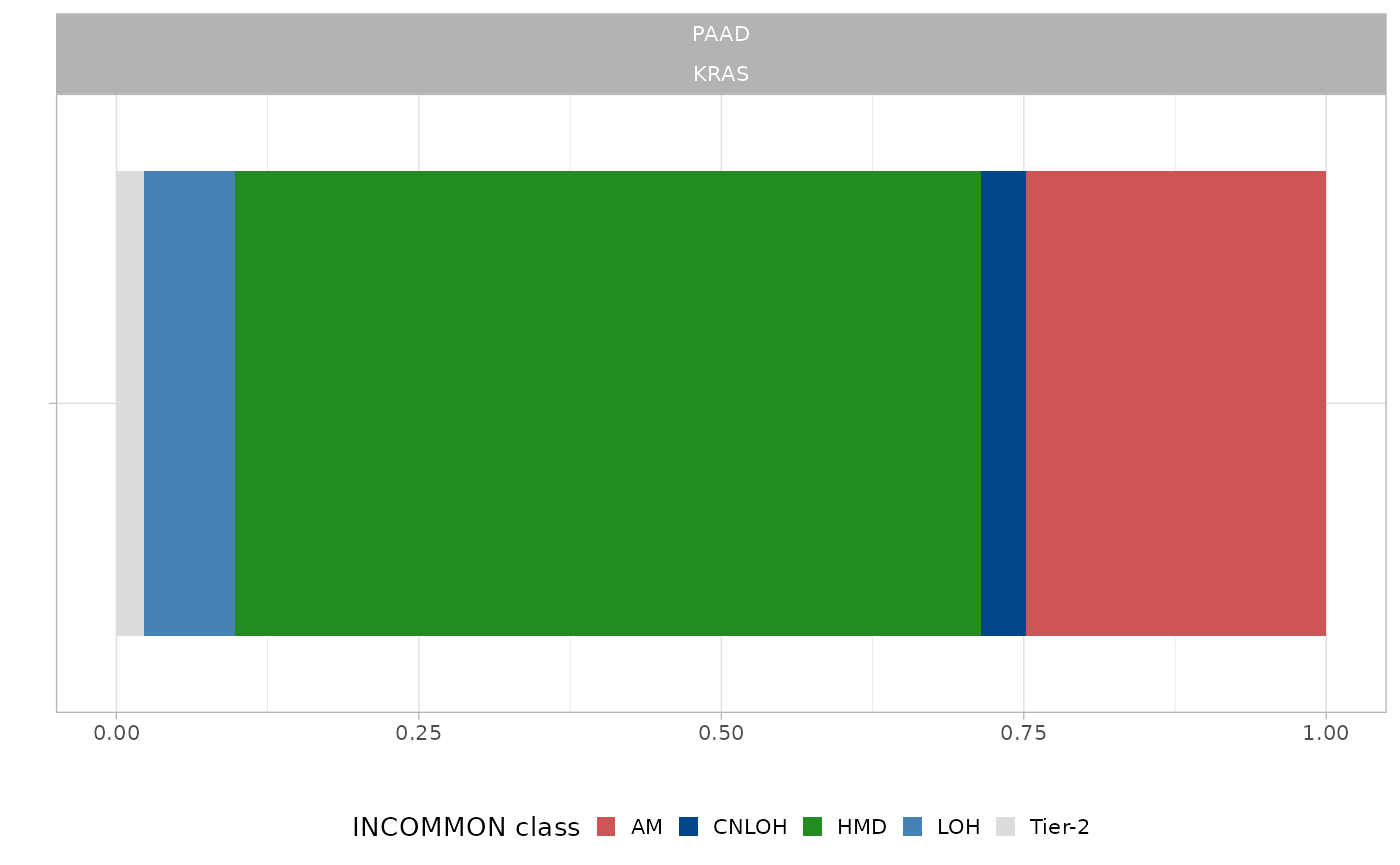
1.3 Visualising priors
The prior distribution used in a fit can be visualised a posteriori
using the internal plotting function plot_prior. We can
plot the prior distribution specific to a gene and tumour type used in
the example classified MSK-MET data.
For example:
data("MSK_classified")
plot_prior(x = MSK_classified,
gene = 'KRAS',
tumor_type = 'PAAD')
#> ✔ Loading CNAqc, 'Copy Number Alteration quality check'. Support : <https://caravagn.github.io/CNAqc/>