devil is an R package for differential expression
analysis in single-cell RNA sequencing (scRNA-seq) data. It supports
both single- and multi-patient experimental designs, implementing robust
statistical methods to identify differentially expressed genes while
accounting for technical and biological variability.
Key features are:
- Flexible experimental design support (single/multiple patients)
- Robust statistical testing framework
- Efficient implementation for large-scale datasets
Installation
You can install the current version of devil from GitHub with:
devtools::install_github("caravagnalab/devil")Example
This tutorial walks through a minimal, end-to-end workflow for
differential expression (DE) with devil on
a public scRNA-seq dataset. You will: (1) load data, (2) filter
cells/genes, (3) build a design, (4) fit the model, (5) specify
contrasts, and (6) visualize results.
If your study has multiple patients/donors, devil can
compute clustered (patient-aware) standard errors via a
cluster argument.
Prerequisites
# If needed:
# install.packages("BiocManager")
# BiocManager::install(c("scRNAseq","SingleCellExperiment"))
library(devil)
library(scRNAseq)
#> Loading required package: SingleCellExperiment
#> Loading required package: SummarizedExperiment
#> Loading required package: MatrixGenerics
#> Loading required package: matrixStats
#>
#> Attaching package: 'MatrixGenerics'
#> The following objects are masked from 'package:matrixStats':
#>
#> colAlls, colAnyNAs, colAnys, colAvgsPerRowSet, colCollapse,
#> colCounts, colCummaxs, colCummins, colCumprods, colCumsums,
#> colDiffs, colIQRDiffs, colIQRs, colLogSumExps, colMadDiffs,
#> colMads, colMaxs, colMeans2, colMedians, colMins, colOrderStats,
#> colProds, colQuantiles, colRanges, colRanks, colSdDiffs, colSds,
#> colSums2, colTabulates, colVarDiffs, colVars, colWeightedMads,
#> colWeightedMeans, colWeightedMedians, colWeightedSds,
#> colWeightedVars, rowAlls, rowAnyNAs, rowAnys, rowAvgsPerColSet,
#> rowCollapse, rowCounts, rowCummaxs, rowCummins, rowCumprods,
#> rowCumsums, rowDiffs, rowIQRDiffs, rowIQRs, rowLogSumExps,
#> rowMadDiffs, rowMads, rowMaxs, rowMeans2, rowMedians, rowMins,
#> rowOrderStats, rowProds, rowQuantiles, rowRanges, rowRanks,
#> rowSdDiffs, rowSds, rowSums2, rowTabulates, rowVarDiffs, rowVars,
#> rowWeightedMads, rowWeightedMeans, rowWeightedMedians,
#> rowWeightedSds, rowWeightedVars
#> Loading required package: GenomicRanges
#> Loading required package: stats4
#> Loading required package: BiocGenerics
#> Loading required package: generics
#>
#> Attaching package: 'generics'
#> The following objects are masked from 'package:base':
#>
#> as.difftime, as.factor, as.ordered, intersect, is.element, setdiff,
#> setequal, union
#>
#> Attaching package: 'BiocGenerics'
#> The following objects are masked from 'package:stats':
#>
#> IQR, mad, sd, var, xtabs
#> The following objects are masked from 'package:base':
#>
#> anyDuplicated, aperm, append, as.data.frame, basename, cbind,
#> colnames, dirname, do.call, duplicated, eval, evalq, Filter, Find,
#> get, grep, grepl, is.unsorted, lapply, Map, mapply, match, mget,
#> order, paste, pmax, pmax.int, pmin, pmin.int, Position, rank,
#> rbind, Reduce, rownames, sapply, saveRDS, table, tapply, unique,
#> unsplit, which.max, which.min
#> Loading required package: S4Vectors
#>
#> Attaching package: 'S4Vectors'
#> The following object is masked from 'package:utils':
#>
#> findMatches
#> The following objects are masked from 'package:base':
#>
#> expand.grid, I, unname
#> Loading required package: IRanges
#> Loading required package: Seqinfo
#> Loading required package: Biobase
#> Welcome to Bioconductor
#>
#> Vignettes contain introductory material; view with
#> 'browseVignettes()'. To cite Bioconductor, see
#> 'citation("Biobase")', and for packages 'citation("pkgname")'.
#>
#> Attaching package: 'Biobase'
#> The following object is masked from 'package:MatrixGenerics':
#>
#> rowMedians
#> The following objects are masked from 'package:matrixStats':
#>
#> anyMissing, rowMedians
library(SingleCellExperiment)
library(SummarizedExperiment)
library(Matrix)
#>
#> Attaching package: 'Matrix'
#> The following object is masked from 'package:S4Vectors':
#>
#> expand
library(dplyr)
#>
#> Attaching package: 'dplyr'
#> The following object is masked from 'package:Biobase':
#>
#> combine
#> The following objects are masked from 'package:GenomicRanges':
#>
#> intersect, setdiff, union
#> The following object is masked from 'package:Seqinfo':
#>
#> intersect
#> The following objects are masked from 'package:IRanges':
#>
#> collapse, desc, intersect, setdiff, slice, union
#> The following objects are masked from 'package:S4Vectors':
#>
#> first, intersect, rename, setdiff, setequal, union
#> The following objects are masked from 'package:BiocGenerics':
#>
#> combine, intersect, setdiff, setequal, union
#> The following object is masked from 'package:generics':
#>
#> explain
#> The following object is masked from 'package:matrixStats':
#>
#> count
#> The following objects are masked from 'package:stats':
#>
#> filter, lag
#> The following objects are masked from 'package:base':
#>
#> intersect, setdiff, setequal, unionLoad and inspect data
We’ll use the Baron pancreas dataset from scRNAseq.
sce <- scRNAseq::BaronPancreasData() # SingleCellExperiment
sce
#> class: SingleCellExperiment
#> dim: 20125 8569
#> metadata(0):
#> assays(1): counts
#> rownames(20125): A1BG A1CF ... ZZZ3 pk
#> rowData names(0):
#> colnames(8569): human1_lib1.final_cell_0001 human1_lib1.final_cell_0002
#> ... human4_lib3.final_cell_0700 human4_lib3.final_cell_0701
#> colData names(2): donor label
#> reducedDimNames(0):
#> mainExpName: NULL
#> altExpNames(0):Extract counts and metadata using accessors:
counts <- SummarizedExperiment::assay(sce, "counts")
meta <- as.data.frame(SummarizedExperiment::colData(sce))
cat("Genes:", nrow(counts), "\nCells:", ncol(counts), "\n")
#> Genes: 20125
#> Cells: 8569
stopifnot("label" %in% colnames(meta))
head(meta$label)
#> [1] "acinar" "acinar" "acinar" "acinar" "acinar" "acinar"Tip: If you have a patient/donor column (often
donor or patient), keep it, we’ll optionally
pass it to cluste= later.
Light filtering
Keep the three most abundant cell types; filter lowly expressed genes.
# keep 3 largest cell types
top3 <- names(sort(table(meta$label), decreasing = TRUE))[1:3]
keep_cells <- meta$label %in% top3
counts <- counts[, keep_cells, drop = FALSE]
meta <- meta[keep_cells, , drop = FALSE]
# gene filter: expressed (>=1 UMI) in >= 1% of kept cells
min_cells <- max(1, floor(0.01 * ncol(counts)))
keep_genes <- Matrix::rowSums(counts >= 1) >= min_cells
counts <- counts[keep_genes, , drop = FALSE]
cat("After filtering — Genes:", nrow(counts), "Cells:", ncol(counts), "\n")
#> After filtering — Genes: 11951 Cells: 5928
table(meta$label)
#>
#> alpha beta ductal
#> 2326 2525 1077Optionally restrict to highly expressed genes for a faster demo (skip for real analyses):
Design matrix
Build a no-intercept design so each coefficient corresponds to a cell type.
meta$label <- droplevels(factor(meta$label))
design <- model.matrix(~ 0 + label, data = meta)
colnames(design) <- gsub("^label", "", colnames(design))
colnames(design)
#> [1] "alpha" "beta" "ductal"(Optional) Cluster variable for patient-aware SEs, if available:
Fit the model
fit_devil() expects a counts matrix (genes × cells), a
design (cells × covariates). The parameters
size_factors="normed_sum" computes internally a size factor
that will scale expression based on the library size of each cell.
fit <- devil::fit_devil(
input_matrix = as.matrix(counts),
design_matrix = design,
overdispersion = "MOM",
offset = 1e-6,
init_overdispersion = NULL,
size_factors = "normed_sum",
parallel.cores = 1,
verbose = TRUE,
max_iter = 200,
tolerance = 1e-4
)
#> Compute size factors
#> Calculating size factors using method: normed_sum
#> Size factors calculated successfully.
#> Range: [0.1036, 8.0148]
#> Initialize theta
#> Initialize beta
#> Fitting beta coefficients
#> Fit overdispersion (mode = MOM)Specify contrasts
With a no-intercept design, each column is a cell-type mean on the
log scale.
To test “beta vs ductal”, define the contrast
(+1 * beta) + (-1 * ductal) and zero elsewhere.
make_contrast <- function(design, from, to) {
stopifnot(from %in% colnames(design), to %in% colnames(design))
c <- rep(0, ncol(design))
names(c) <- colnames(design)
c[from] <- 1
c[to] <- -1
as.numeric(c)
}
contrast <- make_contrast(design, from = "beta", to = "ductal")
contrast
#> [1] 0 1 -1If your labels differ, update from/to
accordingly—use colnames(design) to see available
levels.
Test for differential expression
Run a Wald test with optional clustered SEs if
cluster exists.
test <- devil::test_de(
fit,
contrast = contrast,
max_lfc = 20, # Cap extreme fold changes
cluster = cluster # NULL if not present; enables patient-aware SE if provided
)
#> Converting clusters to numeric factors
# Add gene names if missing
if (!("name" %in% colnames(test))) {
if (!is.null(rownames(counts))) {
test$name <- rownames(counts)
} else {
test$name <- as.character(seq_len(nrow(test)))
}
}Quick peek at the top hits:
test %>%
dplyr::arrange(adj_pval, desc(abs(lfc))) %>%
dplyr::select(name, lfc, pval, adj_pval) %>%
head(10)
#> # A tibble: 10 × 4
#> name lfc pval adj_pval
#> <chr> <dbl> <dbl> <dbl>
#> 1 KRT7 -9.35 5.43e-212 2.72e-209
#> 2 CPE 4.40 7.38e-171 1.85e-168
#> 3 HADH 3.32 6.30e-150 1.05e-147
#> 4 CD24 -3.77 6.80e-136 8.50e-134
#> 5 PEMT 3.95 1.37e-126 1.37e-124
#> 6 PMEPA1 -7.03 1.63e-124 1.36e-122
#> 7 TACSTD2 -8.83 2.52e-117 1.80e-115
#> 8 S100A10 -6.08 5.73e-111 3.58e-109
#> 9 UCHL1 4.63 1.39e-107 7.72e-106
#> 10 TMOD1 4.67 5.65e-103 2.82e-101Visualize results
devil::plot_volcano(
test,
lfc_cut = 1,
pval_cut = 0.05,
labels = TRUE,
point_size = 1.8,
title = "beta vs ductal"
)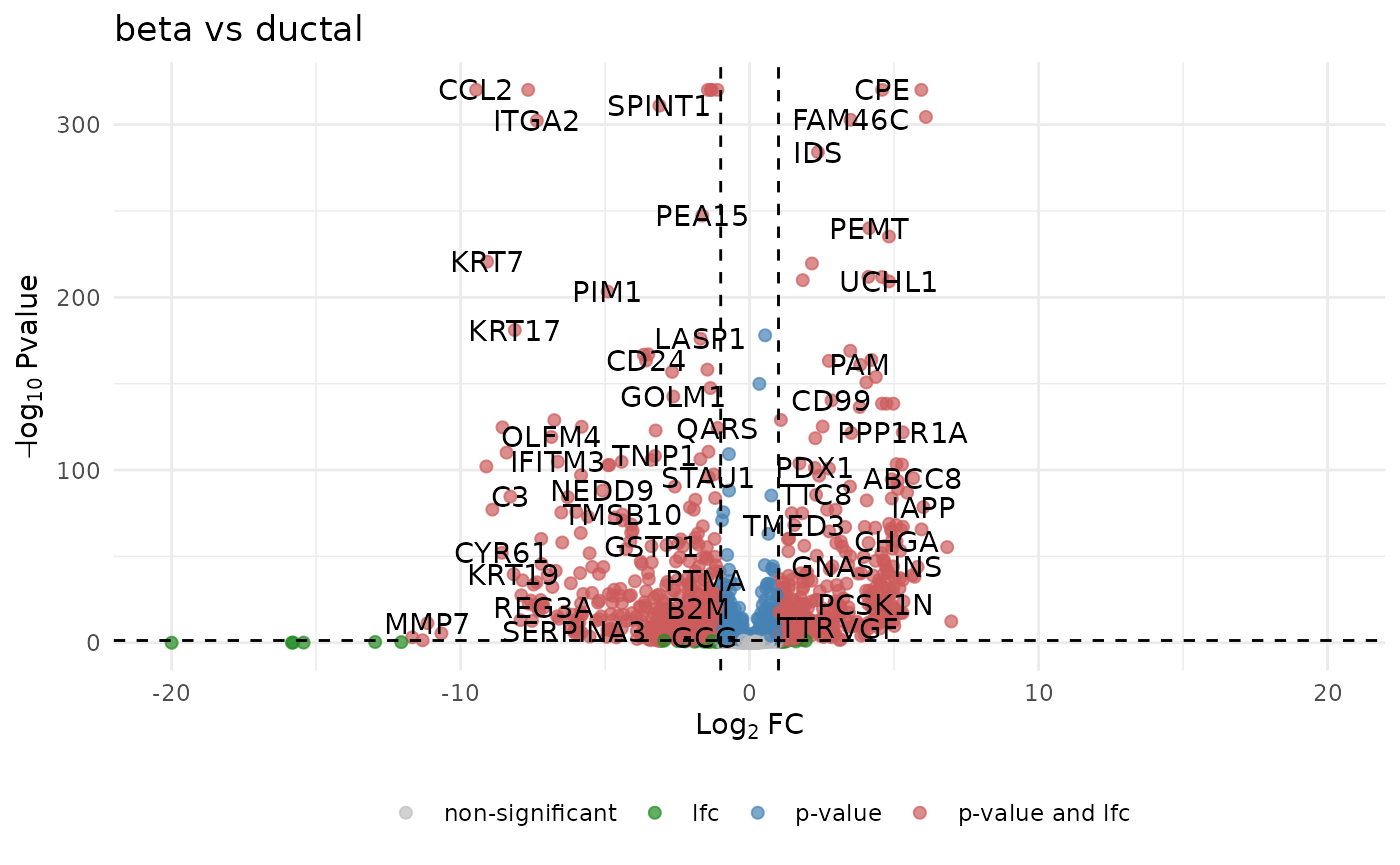
Volcano plot of DE genes
Session info
sessionInfo()
#> R version 4.5.2 (2025-10-31)
#> Platform: x86_64-pc-linux-gnu
#> Running under: Ubuntu 24.04.3 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
#>
#> locale:
#> [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
#> [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
#> [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
#> [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: UTC
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats4 stats graphics grDevices utils datasets methods
#> [8] base
#>
#> other attached packages:
#> [1] dplyr_1.1.4 Matrix_1.7-4
#> [3] scRNAseq_2.24.0 SingleCellExperiment_1.32.0
#> [5] SummarizedExperiment_1.40.0 Biobase_2.70.0
#> [7] GenomicRanges_1.62.1 Seqinfo_1.0.0
#> [9] IRanges_2.44.0 S4Vectors_0.48.0
#> [11] BiocGenerics_0.56.0 generics_0.1.4
#> [13] MatrixGenerics_1.22.0 matrixStats_1.5.0
#> [15] devil_0.99.0
#>
#> loaded via a namespace (and not attached):
#> [1] DBI_1.2.3 bitops_1.0-9
#> [3] httr2_1.2.2 rlang_1.1.6
#> [5] magrittr_2.0.4 gypsum_1.6.0
#> [7] compiler_4.5.2 RSQLite_2.4.5
#> [9] DelayedMatrixStats_1.32.0 GenomicFeatures_1.62.0
#> [11] png_0.1-8 systemfonts_1.3.1
#> [13] vctrs_0.6.5 ProtGenerics_1.42.0
#> [15] pkgconfig_2.0.3 crayon_1.5.3
#> [17] fastmap_1.2.0 dbplyr_2.5.1
#> [19] XVector_0.50.0 labeling_0.4.3
#> [21] utf8_1.2.6 Rsamtools_2.26.0
#> [23] rmarkdown_2.30 UCSC.utils_1.6.1
#> [25] ragg_1.5.0 bit_4.6.0
#> [27] xfun_0.55 cachem_1.1.0
#> [29] cigarillo_1.0.0 GenomeInfoDb_1.46.2
#> [31] jsonlite_2.0.0 blob_1.2.4
#> [33] rhdf5filters_1.22.0 DelayedArray_0.36.0
#> [35] Rhdf5lib_1.32.0 BiocParallel_1.44.0
#> [37] parallel_4.5.2 R6_2.6.1
#> [39] RColorBrewer_1.1-3 bslib_0.9.0
#> [41] rtracklayer_1.70.0 jquerylib_0.1.4
#> [43] Rcpp_1.1.0 knitr_1.50
#> [45] tidyselect_1.2.1 abind_1.4-8
#> [47] yaml_2.3.12 codetools_0.2-20
#> [49] curl_7.0.0 lattice_0.22-7
#> [51] alabaster.sce_1.10.0 tibble_3.3.0
#> [53] S7_0.2.1 withr_3.0.2
#> [55] KEGGREST_1.50.0 evaluate_1.0.5
#> [57] desc_1.4.3 BiocFileCache_3.0.0
#> [59] alabaster.schemas_1.10.0 ExperimentHub_3.0.0
#> [61] Biostrings_2.78.0 pillar_1.11.1
#> [63] BiocManager_1.30.27 filelock_1.0.3
#> [65] RCurl_1.98-1.17 ggplot2_4.0.1
#> [67] BiocVersion_3.22.0 ensembldb_2.34.0
#> [69] scales_1.4.0 sparseMatrixStats_1.22.0
#> [71] alabaster.base_1.10.0 glue_1.8.0
#> [73] alabaster.ranges_1.10.0 alabaster.matrix_1.10.0
#> [75] lazyeval_0.2.2 tools_4.5.2
#> [77] AnnotationHub_4.0.0 BiocIO_1.20.0
#> [79] GenomicAlignments_1.46.0 fs_1.6.6
#> [81] XML_3.99-0.20 rhdf5_2.54.1
#> [83] grid_4.5.2 AnnotationDbi_1.72.0
#> [85] HDF5Array_1.38.0 restfulr_0.0.16
#> [87] cli_3.6.5 rappdirs_0.3.3
#> [89] textshaping_1.0.4 S4Arrays_1.10.1
#> [91] AnnotationFilter_1.34.0 gtable_0.3.6
#> [93] alabaster.se_1.10.0 sass_0.4.10
#> [95] digest_0.6.39 SparseArray_1.10.7
#> [97] farver_2.1.2 rjson_0.2.23
#> [99] memoise_2.0.1 htmltools_0.5.9
#> [101] pkgdown_2.2.0 lifecycle_1.0.4
#> [103] h5mread_1.2.1 httr_1.4.7
#> [105] bit64_4.6.0-1