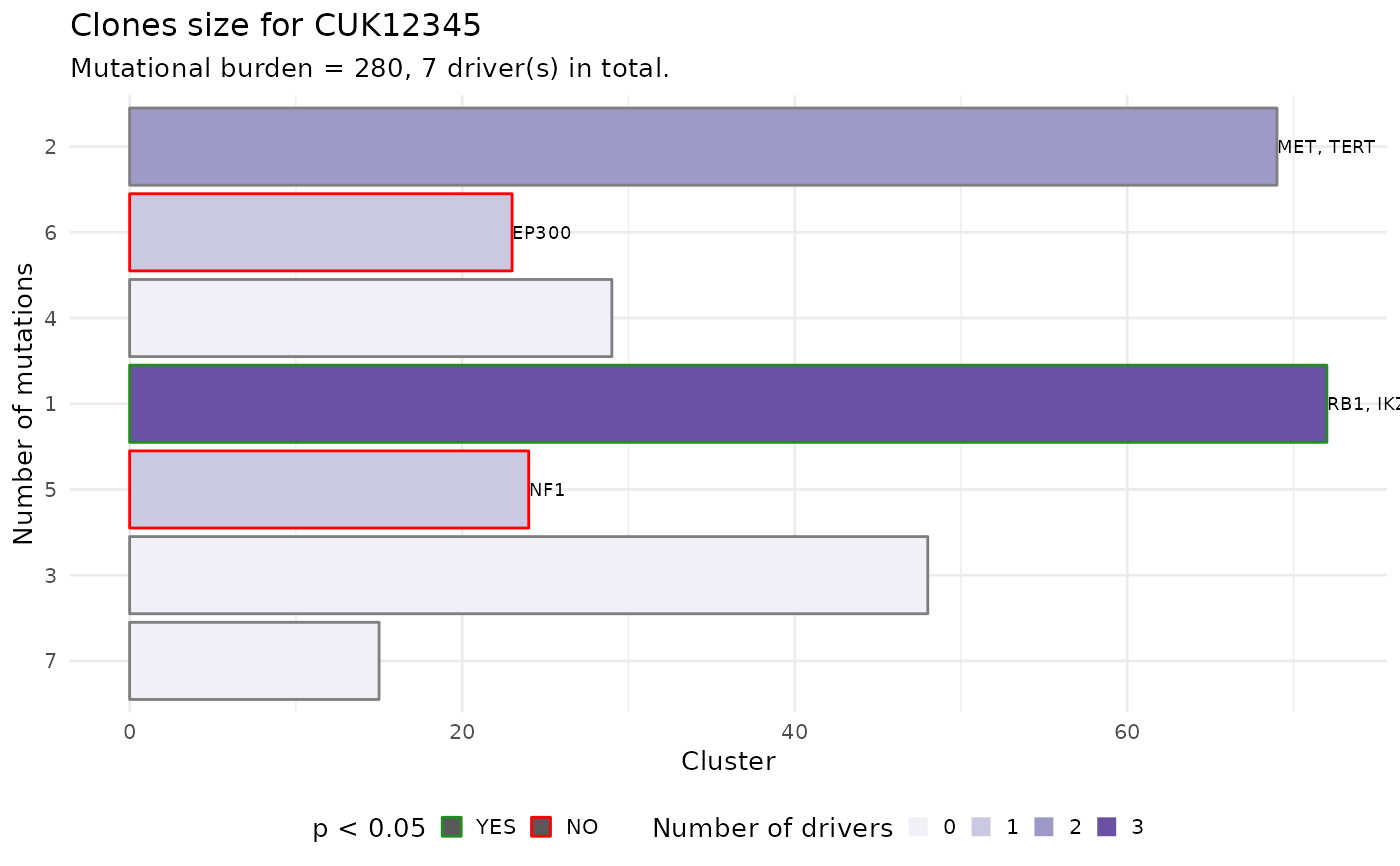
Plot a clone size histogram, and test for.
plot_clone_size.RdThis function creates a ggplot
barplot of the clone size values foe each clone in the patient's data.
The size of a clone is defined as the number of mutations assigned
to it, and is provided in input.
The barplot is annotated to report wether a subclone with a driver is significantly larger than the expected size for a subclone without driver. To carry out this test subclones without drivers are used to estimate the parameters of a univariate Gaussian distribution (mean and standard deviation), the p-value is then computed from the fit distribution through the `pnorm` function.
The confidence level for the test can be passed as parameter.
Examples
data('ctree_input')
x = ctrees(
ctree_input$CCF_clusters,
ctree_input$drivers,
ctree_input$samples,
ctree_input$patient,
ctree_input$sspace.cutoff,
ctree_input$n.sampling,
ctree_input$store.max
)
#> [ ctree ~ clone trees generator for CUK12345 ]
#>
#> # A tibble: 7 × 7
#> cluster nMuts is.driver is.clonal R1 R2 R3
#> <chr> <int> <lgl> <lgl> <dbl> <dbl> <dbl>
#> 1 1 72 TRUE FALSE 0 0.92 0
#> 2 2 69 TRUE TRUE 0.99 0.98 0.99
#> 3 3 48 FALSE FALSE 0 0 0.49
#> 4 4 29 FALSE FALSE 0.01 0.01 0.93
#> 5 5 24 TRUE FALSE 0.78 0 0
#> 6 6 23 TRUE FALSE 0.98 0.03 0.98
#> 7 7 15 FALSE FALSE 0 0.41 0
#>
#> ✔ Trees per region 1, 3, 1
#> ℹ Total 3 tree structures - search is exahustive
#>
#> ── Ranking trees
#> ✔ 3 trees with non-zero score, storing 3
plot_clone_size(x[[1]])
#> Warning: Removed 3 rows containing missing values or values outside the scale range
#> (`geom_text()`).